
20-F: Annual and transition report of foreign private issuers pursuant to Section 13 or 15(d)
Published on April 1, 2019
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 20-F
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended December 31, 2018
Commission File No.: 001-38370
CollPlant Holdings Ltd.
(Exact name of registrant as specified in its charter)
Translation of registrant’s name into English: Not applicable
| State of Israel |
4 Oppenheimer, Weizmann Science Park Rehovot 7670104 , Israel Tel: +972 73 232 5600 |
|
| (Jurisdiction of incorporation or organization) | (Address of principal executive offices) |
Yehiel Tal
Chief Executive Officer
+972 73 232 5600
Yehiel@CollPlant.com
4 Oppenheimer, Weizmann Science Park
Rehovot 7670104 , Israel
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act:
| Title of each class to be registered |
Name of each exchange on which each class is to be registered |
|
| American Depositary Shares, each representing fifty (50) ordinary shares, par value NIS 0.03 per share | The Nasdaq Stock Market LLC | |
| Ordinary shares, par value NIS 0.03 per share* | The Nasdaq Stock Market LLC* |
* Not for trading, but only in connection with the registration of the American Depositary Shares pursuant to requirements of the Securities and Exchange Commission.
Securities registered or to be registered pursuant to Section 12(g) of the Act: None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act: None
Number of outstanding shares of each of the issuer’s classes of capital or common stock as of December 31, 2018: 190,735,668 ordinary shares.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ☐ No ☒
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Exchange Act of 1934.
Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Exchange Act during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T during the preceding 12 months.
Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or an emerging growth company.
| Large accelerated filer | ☐ | Accelerated filer | ☐ | Non-accelerated filer | ☒ | |
| Emerging Growth Company | ☒ |
If an emerging growth company that prepares its financial statements in accordance with U.S. GAAP, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of the Securities Act. ☐
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing.
U.S. GAAP ☐
International Financial Reporting Standards as issued by the International Accounting Standards Board ☒
Other ☐
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow.
☐ Item 17 ☐ Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company.
Yes ☐ No ☒
i
ii
We are a regenerative medicine company focused on developing and commercializing tissue repair products, initially for 3D-bio printing of tissues and organs, dermal fillers for aesthetics, orthobiologics, and advanced wound care markets. Our products are based on our recombinant type I human collagen, or rhCollagen, a form of human collagen produced with our proprietary plant based genetic engineering technology.
On January 31, 2018, our American Depositary Shares, or ADSs, each representing fifty of our ordinary shares commenced trading on the Nasdaq Capital Market under the symbols “CLGN”. Our ADSs were quoted on the OTCQX from March 2015 to May 25, 2017, and, prior to listing on the Nasdaq Capital Market, quoted on the OTCQB from May 26, 2017 to January 30, 2018. We delisted our ordinary shares from the Tel Aviv Stock Exchange or TASE, and the last date of trading of our ordinary shares was on October 29, 2018.
Unless the context requires otherwise, the terms “CollPlant,” “we,” “us,” “our,” “the Company,” and similar designations refer to CollPlant Holdings Ltd. and its wholly owned subsidiary CollPlant Ltd. References to “ordinary shares”, “ADSs”, “warrants” and “share capital” refer to the ordinary shares, ADSs, warrants and share capital, respectively, of CollPlant.
References to “U.S. dollars” and “$” are to currency of the United States of America, and references to “NIS” are to New Israeli Shekels. References to “ordinary shares” are to our ordinary shares, par value NIS 0.03 per share. We report financial information under International Financial Reporting Standards, or IFRS, as issued by the International Accounting Standards Board, or IASB, and none of the financial statements were prepared in accordance with generally accepted accounting principles in the United States.
Unless otherwise indicated, U.S. dollar translations of NIS amounts presented in this annual report on Form 20-F for the year ended on December 31, 2018 are translated using the rate of NIS 3.748 to $1.00, the exchange rate reported by the Bank of Israel on December 31, 2018, U.S. dollar translations of NIS amounts presented in this annual report on Form 20-F for the year ended on December 31, 2017 are translated using the rate of NIS 3.467 to $1.00, the exchange rate reported by the Bank of Israel on December 31, 2017, and U.S. dollar translations of NIS amounts presented in this annual report on Form 20-F for the year ended on December 31, 2016 are translated using the rate of NIS 3.845 to $1.00, the exchange rate reported by the Bank of Israel on December 31, 2016.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
Certain information included or incorporated by reference in this Annual Report on Form 20-F may be deemed to be “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995 and other securities laws. Forward-looking statements are often characterized by the use of forward-looking terminology such as “may,” “will,” “expect,” “anticipate,” “estimate,” “continue,” “believe,” “should,” “intend,” “project” or other similar words, but are not the only way these statements are identified.
These forward-looking statements may include, but are not limited to, statements relating to our objectives, plans and strategies, statements that contain projections of results of operations or of financial condition, expected capital needs and expenses, statements relating to the research, development, completion and use of our products, and all statements (other than statements of historical facts) that address activities, events or developments that we intend, expect, project, believe or anticipate will or may occur in the future.
Forward-looking statements are not guarantees of future performance and are subject to risks and uncertainties. We have based these forward-looking statements on assumptions and assessments made by our management in light of their experience and their perception of historical trends, current conditions, expected future developments and other factors they believe to be appropriate.
iii
Important factors that could cause actual results, developments and business decisions to differ materially from those anticipated in these forward-looking statements include, among other things:
| ● | our history of significant losses, our ability to continue as a going concern, and our need to raise additional capital and our inability to obtain additional capital on acceptable terms, or at all; |
| ● | our expectations regarding the timing and cost of commencing clinical trials with respect to tissues and organs which are based on our rhCollagen based BioInk, dermal fillers for aesthetics, VergenixSTR, and VergenixFG; | |
| ● | our ability to obtain favorable pre-clinical and clinical trial results; |
| ● | regulatory action with respect to rhCollagen based BioInk, dermal fillers for aesthetics, VergenixSTR, and VergenixFG including but not limited to acceptance of an application for marketing authorization, review and approval of such application, and, if approved, the scope of the approved indication and labeling; |
| ● | commercial success and market acceptance of our rhCollagen based BioInk, dermal fillers for aesthetics, VergenixSTR, and VergenixFG; |
| ● | our ability to establish sales and marketing capabilities or enter into agreements with third parties and our reliance on third party distributors and resellers; |
| ● | our ability to establish and maintain strategic partnerships and other corporate collaborations; |
| ● | our reliance on third parties to conduct some or all aspects of our product manufacturing; |
| ● | the scope of protection we are able to establish and maintain for intellectual property rights and our ability to operate our business without infringing the intellectual property rights of others; |
| ● | the overall global economic environment; |
| ● | the impact of competition and new technologies; |
| ● | general market, political, and economic conditions in the countries in which we operate; | |
| ● | projected capital expenditures and liquidity; | |
| ● | changes in our strategy; |
| ● | litigation and regulatory proceedings; and |
| ● | those factors referred to in “Item 3.D. Risk Factors,” “Item 4. Information on the Company,” and “Item 5. Operating and Financial Review and Prospects”, as well as in this annual report on Form 20-F generally. |
Readers are urged to carefully review and consider the various disclosures made throughout this Annual Report on Form 20-F which are designed to advise interested parties of the risks and factors that may affect our business, financial condition, results of operations and prospects.
You should not put undue reliance on any forward-looking statements. Any forward-looking statements in this annual report on Form 20-F are made as of the date hereof and are expressly qualified in their entirety by the cautionary statements included in this Annual Report. We undertake no obligation to publicly update or revise any forward-looking statements, whether as a result of new information, future events or otherwise, except as required by law.
In addition, the section of this Annual Report on Form 20-F entitled “Item 4. Information on the Company” contains information obtained from independent industry sources and other sources that we have not independently verified.
iv
Market data and certain industry data and forecasts used throughout this Annual Report on Form 20-F were obtained from internal company surveys, market research, consultant surveys commissioned by the Company, publicly available information, reports of governmental agencies and industry publications and surveys. Industry surveys, publications, consultant surveys commissioned by the Company and forecasts generally state that the information contained therein has been obtained from sources believed to be reliable. However, this information may prove to be inaccurate because of the method by which some of the data for the estimates is obtained or because this information cannot always be verified with complete certainty due to the limits on the availability and reliability of raw data, the voluntary nature of the data gathering process and other limitations and uncertainties. As a result, the market and industry data and forecasts included or incorporated by reference in this annual report, and estimates and beliefs based on that data, may not be reliable. We have relied on certain data from third-party sources, including internal surveys, industry forecasts and market research, which we believe to be reliable based on our management’s knowledge of the industry. However, we have not ascertained the underlying economic assumptions relied upon therein. Forecasts are particularly likely to be inaccurate, especially over long periods of time. In addition, we do not necessarily know what assumptions regarding general economic growth were used in preparing the forecasts we cite. Statements as to our market position are based to the best of our knowledge on the most currently available data. While we are not aware of any misstatements regarding the industry data presented in this annual report, our estimates involve risks and uncertainties and are subject to change based on various factors, including those discussed under the heading “Risk Factors” in this Annual Report.
Statements made in this Annual Report on Form 20-F concerning the contents of any agreement, contract or other document are summaries of such agreements, contracts or documents and are not a complete description of all of their terms. If we filed any of these agreements, contracts or documents as exhibits to this Report or to any previous filing with the Securities and Exchange Commission, or SEC, you may read the document itself for a complete understanding of its terms.
v
| ITEM 1. | IDENTITY OF DIRECTORS, SENIOR MANAGEMENT AND ADVISERS |
Not applicable.
| ITEM 2. | OFFER STATISTICS AND EXPECTED TIMETABLE |
Not applicable.
| ITEM 3. | KEY INFORMATION |
| A. | Selected Financial Data |
The following table sets forth our selected consolidated financial data for the periods ended and as of the dates indicated. The following selected consolidated financial data should be read in conjunction with the financial information, “Item 5. Operational and Financial Review and Prospects” and other information provided elsewhere in this Annual Report on Form 20-F and our consolidated financial statements and related notes. The selected consolidated financial data in this section is not intended to replace the consolidated financial statements and is qualified in its entirety thereby.
The selected consolidated statement of comprehensive loss data for the years ended December 31, 2018, 2017 and 2016, and the selected consolidated statement of financial position data as of December 31, 2018 and December 31, 2017, have been derived from our audited consolidated financial statements and notes thereto set forth elsewhere in this Annual Report on Form 20-F. The selected consolidated statement of financial position data as of December 31, 2016 have been derived from our audited consolidated financial statements not included in this Annual Report.
1
Statement of comprehensive loss data
| Year ended December 31, | ||||||||||||||||
| 2016 | 2017 | 2018 | 2018 | |||||||||||||
| (NIS in thousands except per share data) |
(Convenience translation into USD in thousands except per share data(1)) |
|||||||||||||||
| Revenues | 292 | 1,668 | 18,034 | 4,812 | ||||||||||||
| Cost of revenue | — | 52 | 1,426 | 380 | ||||||||||||
| Gross Profit | 292 | 1,616 | 16,608 | 4,432 | ||||||||||||
| Research and development expenses | 29,200 | 16,921 | 16,213 | 4,325 | ||||||||||||
| Participation in research and development expenses | (12,411 | ) | (2,855 | ) | 3,227 | 861 | ||||||||||
| Research and development expenses, net | 16,789 | 14,066 | 19,440 | 5,186 | ||||||||||||
| General, administrative and marketing expenses | 11,048 | 8,303 | 12,480 | 3,330 | ||||||||||||
| Operating loss | (27,545 | ) | (20,753 | ) | (15,312) | (4,084 | ) | |||||||||
| Financial income | 93 | 253 | 1,650 | 440 | ||||||||||||
| Financial expenses | (441) | (380 | ) | (225) | (60) | |||||||||||
| Financial (expenses) income, net | (348) | (127) | 1,425 | 380 | ||||||||||||
| Comprehensive loss | 27,893 | 20,880 | 13,887 | 3,704 | ||||||||||||
| Loss per ordinary share, basic and diluted | 0.28 | 0.16 | 0.06 | 0.02 | ||||||||||||
| Weighted average ordinary shares outstanding, basic and diluted | 100,624,945 | 133,187,048 | 219,229,229 | 219,229,229 | ||||||||||||
| (1) | Calculated using the exchange rate reported by the Bank of Israel for December 31, 2018, at the rate of one U.S. dollar per NIS 3.748. |
Statement of financial position data
| December 31, | ||||||||||||||||
| 2016 | 2017 | 2018 | 2018 | |||||||||||||
|
(NIS in thousands) |
(Convenience translation into USD in thousands(1)) |
|||||||||||||||
| Cash and cash equivalents | 3,797 | 17,817 | 20,065 | 5,354 | ||||||||||||
| Total assets | 14,433 | 28,045 | 34,082 | 9,092 | ||||||||||||
| Total liabilities | 9,273 | 18,962 | 16,156 | 4,309 | ||||||||||||
| Ordinary Shares | 3,207 | 4,998 | 5,716 | 1,525 | ||||||||||||
| Total equity | 5,160 | 9,083 | 17,926 | 4,783 | ||||||||||||
| (1) | Calculated using the exchange rate reported by the Bank of Israel for December 31, 2018 at the rate of one U.S. dollar per NIS 3.748. |
2
| B. | Capitalization and Indebtedness |
Not applicable.
| C. | Reasons for the Offer and Use of Proceeds |
Not applicable.
| D. | Risk Factors |
You should carefully consider the risks described below, together with all of the other information in this Annual Report on Form 20-F. The risks and uncertainties described below are those significant risk factors, currently known and specific to us, that we believe are relevant to an investment in our securities. Additional risks and uncertainties not currently known to us or that we now deem immaterial may also harm us. If any of these risks materialize our business, results of operations or financial condition could suffer, and the price of the ADSs could decline substantially.
Risks Related to Our Financial Position and Capital Requirements
We have incurred significant losses since our inception and anticipate that we will continue to incur significant losses for the foreseeable future.
We are a regenerative medicine company. We have incurred losses in each year since our inception in 2004, including total comprehensive loss of NIS 13.9 million ($3.7 million), NIS 20.9 million ($6.0 million), and NIS 27.9 million ($7.3 million) for the years ended December 31, 2018, December 31, 2017 and December 31, 2016, respectively. As of December 31, 2018, we had an accumulated deficit of NIS 184.7 million ($49.3 million).
We have devoted most of our financial resources to research and development, including our clinical and preclinical development activities. To date, we have financed our operations primarily through the sale of equity securities, grants from government authorities and proceeds from strategic collaborators. The amount of our future net losses will depend, in part, on the rate of our future expenditures. If and when we obtain regulatory approval to market any of our products, our future revenues will depend upon the size of any markets in which our products have received approval, and our ability to achieve sufficient market acceptance, reimbursement from third-party payors and adequate market share for our products in those markets.
3
We expect to continue to incur significant expenses and operating losses for the foreseeable future. We anticipate that our expenses will increase substantially if and as we:
| ● | continue our research and preclinical and clinical development of our products; |
| ● | initiate additional preclinical, clinical, or other studies for our products; |
| ● | seek marketing approvals for any of our products that successfully complete clinical trials; |
| ● | further develop and expand the manufacturing process for our products; |
| ● | establish a sales, marketing, and distribution infrastructure to commercialize our products for which we may obtain marketing approval; |
| ● | seek to identify and validate additional products; |
| ● | maintain, protect, and expand our intellectual property portfolio; |
| ● | attract and retain skilled personnel; |
| ● | create additional infrastructure to support our operations as a public company; and |
| ● | experience any delays or encounter issues with any of the above. |
The net losses we incur may fluctuate significantly from quarter to quarter and year to year, such that a period-to-period comparison of our results of operations may not be a good indication of our future performance. In any particular quarter or quarters, our operating results could be below the expectations of securities analysts or investors, which could cause our share price to decline.
Our recurring operating losses, negative cash flows and current cash position have raised substantial doubt regarding our ability to continue as a going concern.
We have an accumulated deficit as of December 31, 2018, as well as a history of net losses and negative operating cash flows in recent years. We expect to continue incurring losses and negative cash flows from operations until our products (primarily BioInk) reach commercial profitability. As a result of these expected losses and negative cash flows from operations, along with our current cash position, we do not have sufficient cash to meet our liquidity requirements for the following twelve months. Consequently, there is substantial doubt regarding our ability to continue as a going concern. As a result, our independent registered public accounting firm included a “going concern” explanatory paragraph in its report on our financial statements as of and for the year ended December 31, 2018 with respect to this uncertainty. Our ability to continue as a going concern will require us to obtain additional financing to fund our operations. The perception of our ability to continue as a going concern may make it more difficult for us to obtain financing or obtain financing on favorable terms for the continuation of our operations and could result in the loss of confidence by investors, suppliers and employees. If we are not successful in raising capital through equity offerings, debt financings, collaborations, licensing arrangements or any other means or are not successful in reducing our expenses, we may exhaust our cash resources and will be unable to continue our operations. If we cannot continue as a viable entity, our shareholders would likely lose most or all of their investment in us.
4
We will need to raise additional funding, which may not be available on acceptable terms, or at all. Failure to obtain additional capital when needed may force us to delay, limit, or terminate our product development efforts or other operations.
We are conducting clinical and preclinical development of our products and we intend to continue advancing their development. Developing medical products is expensive, and we expect our research and development expenses to continue to be a material part of our expenses, and may increase substantially in connection with our ongoing activities, particularly as we advance our products in clinical trials.
As of December 31, 2018, our cash and cash equivalents were $5.4 million. We believe that our existing cash and cash equivalents, will enable us to fund our operating expenses and capital expenditure requirements into the first quarter of 2020. However, our operating plan may change as a result of many factors currently unknown to us, and we may need to seek additional funds sooner than planned, through public or private equity or debt financings, government or other third-party funding, marketing and distribution arrangements, and other collaborations, strategic alliances, and licensing arrangements, or a combination of these approaches. We will require additional capital to obtain U.S. Food and Drug Administration, or FDA, approval and commercialize any product that receives regulatory approval. Even if we believe we have sufficient funds for our current or future operating plans, we may seek additional capital if market conditions are favorable or if we have specific strategic considerations.
Any additional fundraising efforts may divert our management from their day-to-day activities, which may compromise our ability to develop and commercialize our products. In addition, we cannot guarantee that future financing will be available in sufficient amounts or on terms acceptable to us, if at all. Moreover, the terms of any financing may adversely affect the holdings or the rights of our shareholders, and the issuance of additional securities, whether equity or debt, by us, or the possibility of such issuance, may cause the market price of our ordinary shares or ADSs to decline. The sale of additional equity or convertible securities would dilute all of our shareholders. The incurrence of indebtedness would result in increased fixed payment obligations, and we may be required to agree to certain restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell, or license intellectual property rights, and other operating restrictions that could adversely impact our ability to conduct our business. We could also be required to seek funds through arrangements with collaborative partners or otherwise at an earlier stage than otherwise would be desirable, and we may be required to relinquish rights to some of our technologies or products or otherwise agree to terms unfavorable to us.
If we are unable to obtain funding on a timely basis, we may be required to significantly curtail, delay, or discontinue one or more of our research or development programs or the commercialization of any products, and we may be unable to expand our operations or otherwise capitalize on our business opportunities, as desired.
We have received and may continue to receive Israeli governmental grants to assist in the funding of our research and development activities. If we lose our funding from these research and development grants, we may encounter difficulties in the funding of future research and development projects and implementing technological improvements, which would harm our operating results.
Through December 31, 2018 we had received an aggregate of $10.0 million in the form of grants from the Israel Innovation Authority, or the IIA (formerly known as the Office of the Chief Scientist of the Ministry of Economy and Industry, or the OCS). The requirements and restrictions for such grants are found in the Encouragement of Research, Development and Technological Innovation in the Industry Law 5744 1984 (formerly known as the Law for the Encouragement of Research and Development in Industry 5744 1984), or the Innovation Law, and the IIA’s rules and guidelines. Under the IIA’s rules and guidelines, royalties of 3% to 6% on the income generated from sales of products and related services developed in whole or in part under IIA programs are payable to the IIA (depending on the type of the Recipient Company—i.e., whether it is a “Small Company”, a “Large Company” or a “Traditional Industrial Company” as such terms are defined in the IIA’s rules and guidelines), up to the total amount of grants received, including annual interest, all as detailed in the IIA’s rules and guidelines.
5
We developed our platform technologies, at least in part, with funds from these grants, and accordingly we are obligated to pay these royalties on sales of any of our current products that achieve regulatory approval. In addition, the Government of Israel may from time to time audit sales of products which it claims incorporate technology funded via the IIA programs and this may lead to additional royalties being payable on additional products. As of December 31, 2018, the maximum royalty amount that would be payable by us, excluding interest, is $8.5 million. As of December 31, 2018, we paid to the IIA $1.2 million in royalties and payments against the approval to use outside the state of Israel in regards to the License, Development and Commercialization Agreement, or the United License Agreement, entered into by CollPlant Ltd., or CollPlant, our wholly-owned subsidiary. For the year ended December 31, 2018, we received grants totaling $589,000 from the IIA. The grants represented 14% of our gross research and development expenditures for the year ended December 31, 2018. Following the full payment of such royalties and interest, there is generally no further liability for royalty payments; however, other restrictions under the IIA’s rules and guidelines, described below under “Risk Factors—The IIA grants we have received for research and development expenditures restrict our ability to manufacture products and transfer know-how outside of Israel and require us to satisfy specified conditions”, will continue to apply even after we have repaid the full amount of royalties on the grants.
These grants have funded some of our personnel, development activities with subcontractors, and other research and development costs and expenses. However, if these grants are not funded in their entirety or if new grants are not awarded in the future, due to, for example, IIA budget constraints or governmental policy decisions, our ability to fund future research and development and implement technological improvements would be impaired, which would negatively impact our ability to develop our products.
The IIA grants we have received for research and development expenditures restrict our ability to manufacture products and transfer IIA funded know-how outside of Israel and require us to satisfy specified conditions.
Our research and development efforts have been financed, in part, through the grants that we have received from the IIA. We, therefore, must comply with the requirements of the Innovation Law and the IIA’s rules and guidelines.
Under the Innovation Law and the IIA’s rules and guidelines, we are generally prohibited from manufacturing products developed under the IIA’s funding outside of the State of Israel without the prior approval of the IIA (such approval is not required for the transfer of less than 10% of the manufacturing capacity in the aggregate, but a mere notification). We may not receive the required approvals for any proposed transfer of manufacturing activities. In general, in addition to the requirement of obtaining approval to manufacture products developed with IIA grants outside of Israel, the royalty repayment rate would increase and we would be required to pay increased royalties, between 120% and 300% of the grants plus annual interest, depending on the manufacturing volume that is performed outside of Israel. This restriction may impair our ability to outsource manufacturing rights abroad.
A company also has the option of declaring in its IIA grant application its intent to exercise a portion of the manufacturing capacity abroad, thus avoiding the need to obtain additional approval following the receipt of the grant.
Additionally, under the Innovation Law and the IIA’s rules and guidelines, we are prohibited from transferring the IIA’s funded know-how and related intellectual property rights outside of the State of Israel, except under limited circumstances and only with the approval of the IIA’s committee. We may not receive the required approvals for any proposed transfer, and even if we receive the required approvals, we may be required to pay the IIA a redemption fee up to a maximum of 600% of the grant amounts plus interest, depending upon the value of the transferred know-how, our research and development expenses, the amount of the IIA’s support, the time of completion of the IIA supported research project and other factors.
6
A transfer of IIA’s funded know-how to an Israeli company also requires the approval of the IIA’s committee, but will not subject the Company to a payment of a redemption fee (we note that there will be an obligation to pay royalties to the IIA from the income of such sale transaction as part of the royalty payment obligation), and approval may only be granted if the recipient abides by the provisions of applicable laws, including the restrictions on the transfer of know-how and the manufacturing rights outside of Israel and the obligation to pay royalties. No assurance can be given that approval to any such transfer, if requested, will be granted.
Recently, the IIA has published new rules and guidelines with respect to the grant of the right to use know-how that was developed using the IIA’s grants to a foreign entity. According to these rules, the grant of a right to a foreign entity to use the IIA’s funded know-how (without entirely expropriating from the IIA-funded company the possibility of using the IIA’s funded know-how) is subject to receipt of the IIA’s prior approval. This approval is subject to payment to the IIA in accordance with the formulas stipulated in these rules.
These restrictions may impair our ability to sell our technology assets or to perform or outsource manufacturing outside of Israel, or otherwise transfer our know-how outside of Israel. These restrictions may also require us to obtain the approval of the IIA for certain actions and transactions and pay additional royalties and other amounts to the IIA. Furthermore, the consideration available to our shareholders in a transaction involving the transfer outside of Israel of know-how developed with IIA funding (such as a merger or similar transaction) may be reduced by any amounts that we are required to pay to the IIA.
If we fail to comply with the requirements of the Innovation Law, we may be required to refund certain grants previously received along with interest and penalties, and we may become subject to criminal proceedings.
In August 2015, a new amendment to the Innovation Law was enacted, or Amendment No. 7, which came into effect on January 1, 2016. Under Amendment No. 7, the IIA has assumed oversight of activity previously subject to OCS’ responsibility. The IIA was granted wide freedom of action, and among other things, the IIA has the authority to amend the requirements and restrictions which were specified in the Innovation Law before Amendment No. 7 came into effect with respect to the ownership of IIA’s funded know-how (including with respect to the restrictions on transfer of the IIA’s funded know-how and manufacturing activities outside of Israel), as well as with respect to royalty payment obligations which apply to companies that receive grants from the IIA. Amendment No. 7 also includes new provisions with respect to sanctions imposed for violations of the Innovation Law. Although the IIA recently published rules which for the most part adopted the principal provisions and restrictions specified in the Innovation Law prior to the effectiveness of Amendment No. 7, as of the date of this Annual Report on Form 20-F, we are unable to assess the effect of any future rules which may be published by the IIA on our business.
We may not be able to correctly estimate or control our future operating expenses, which could lead to cash shortfalls.
Our operating expenses may fluctuate significantly in the future for various reasons, many of which are outside of our control. These reasons may include:
| ● | the time, resources, and expenses required to conduct clinical trials of, seek regulatory approvals for, manufacture, market, and sell our current products and any additional products we may develop; |
| ● | the time, resources, and expenses required to research and develop, conduct clinical trials of, and seek regulatory approvals for additional indications of our current products; |
| ● | the costs of preparing, filing, prosecuting, defending, and enforcing patent claims and other patent-related costs, including litigation costs or the results of such litigation; |
| ● | any product liability or other lawsuits related to our products and the costs associated with defending them or the results of such lawsuits; |
| ● | the costs to attract and retain personnel with the skills required for effective operations; and |
| ● | the costs associated with being a public company in the United States. |
7
It is difficult to forecast our future performance, which may cause our financial results to fluctuate unpredictably.
Because we do not yet have an established commercial operating history, and because the market for our products may rapidly evolve, it is hard for us to predict our future performance. A number of factors, many of which are outside of our control, may contribute to fluctuations in our financial results assuming that we receive marketing authorizations and begin selling our products. These factors may include variations in:
| ● | market demand for, and acceptance of, our products; |
| ● | our ability to obtain or maintain regulatory approvals; |
| ● | our sales and marketing operations, or the effectiveness of these operations; |
| ● | performance of our third-party contractors; |
| ● | the availability of procedures or products that compete with our products; |
| ● | media coverage of our technologies, the procedures or products of our competitors or our industry; and |
| ● | general economic and political conditions, including changes in general consumer confidence. |
There are uncertainties and risks relating to the Alpha, Meitav Dash and Ami Sagy transactions.
In 2017, we entered into securities purchase agreements with Alpha, Meitav Dash and Ami Sagy providing for up to $7.4 million of financing. The foregoing purchase agreements contain certain covenants that may impact our ability to raise funds in the future, including without limitation, restrictions on future issuances of securities, pre-emption rights and full-ratchet anti-dilution protection.
In addition, the staff of the Israel Securities Authority, or ISA, has informed us that the financings of Meitav Dash and Ami Sagy should be viewed as constituting a single transaction under the provisions of section 270(5)(b)(3) of the Israeli Companies Law, 5759-1999, or the Companies Law. This position is based on several arguments, including the identical price in these financings, their proximity in timing, the similar structure of the financings, including their dates of completion as well as the conditioning of the second closing of Meitav Dash upon the raising of NIS 3.7 million by us, which is an identical amount to the consideration paid by Ami Sagy, and the disclosure with respect to which was published one business day following the disclosure of the Meitav Dash financing. In addition, the Israel Securities Authority, or the ISA, informed us that the Meitav Dash and Ami Sagy financings should be submitted for approval at a general meeting of shareholders as required by Section 270(5) of the Companies Law, since it cannot be determined that the terms of these financings have been set at market terms, considering, among other things, the discount rate embedded in these financings, which was set at 27%-33%. Their position is based on the fact that in our case, there is difficulty in determining market terms which derives, according to the ISA’s position, from the differences in the prevalent discount rates of companies with similar characteristics as us. The differences include the terms of issuance and certain adjustments, including protection for decrease in share price (full ratchet anti-dilution) and the calculation of the fair market value of the warrants. The ISA’s position is that such differences may have implications on the calculation of the discount rates in the Meitav Dash and Ami Sagy financings. Nevertheless, our board of directors believes that in the first place, the said financings should not be considered as a single transaction, and that even if they were viewed as a single transaction, they were entered into on market terms. Accordingly, we have proceeded to complete all closings under the Meitav Purchase Agreement and the Sagy Purchase Agreement without seeking shareholder approval. As a result of the position adopted by the ISA, there is a possibility of derivative claims and class action litigation being brought against us. Such litigation, if instituted, could result in the voiding of the transactions, incurrence of damages, substantial costs and diversion of management attention and resources. In addition, plaintiffs may seek to obtain an injunction prohibiting us from completing the remaining closings on the agreed upon terms, which could prevent the remaining closings from taking place, or from taking place within the expected timeframe. Any conclusion of these matters in a manner adverse to us could have a material adverse effect on our liquidity, financial condition and results of operations.
8
Risks Related to Commercialization of Our Products
The commercial success of any current or future product, if approved, will depend upon the degree of market acceptance by physicians, patients, third-party payors, pharma companies and others in the medical community.
Even if we obtain the requisite regulatory approvals, the commercial success of our products will depend in part on physicians, patients, third party payors, pharma companies and others in the medical community accepting our products as medically useful, cost-effective, and safe. Any product that we bring to the market may not gain market acceptance by physicians, patients, third-party payors, and others in the medical community. If these products do not achieve an adequate level of acceptance, we may not generate significant product revenue and may not become profitable. The degree of market acceptance of these products, if approved for commercial sale, will depend on a number of factors, including:
| ● | the cost, safety, efficacy, and convenience of our products in relation to alternative treatments and products; |
| ● | the ability of third parties to enter into relationships with us without violating their existing agreements; |
| ● | the effectiveness of our sales and marketing efforts; |
| ● | the prevalence and severity of any side effects, including any limitations or warnings contained in a product’s approved labeling; |
| ● | the prevalence and severity of any side effects resulting from the procedure by which our products are administered; |
| ● | the willingness of the target patient population to try new therapies and of physicians to prescribe these therapies; |
| ● | the strength of marketing and distribution support for, and timing of market introduction of, competing products; |
| ● | publicity concerning our products or competing products and treatments; and |
| ● | sufficient third-party insurance coverage or reimbursement. |
Even if a potential product displays a favorable safety and efficacy profile in clinical trials, market acceptance of the product will not be known until after it is launched. Our efforts to educate the medical community and third-party payors on the benefits of the products may require significant resources and may never be successful. Such efforts to educate the marketplace may require more resources than are required by conventional technologies.
We have only limited clinical data to support VergenixFG and VergenixSTR, which may make physicians, patients, third-party payors, and others in the medical community reluctant to accept or purchase our products.
Physicians, patients, third party payors, and others in the medical community will only accept or purchase our products if they believe them to be safe and effective, with advantages over competing products or procedures. To date, we have collected only limited clinical data with which to assess the clinical and economic value of VergenixFG and VergenixSTR. The collection of clinical and economic data and the process of generating peer review publications in support of our product and procedure is an ongoing focus for us. If future publications of clinical studies indicate that procedures using the VergenixFG and VergenixSTR are less safe or less effective than competing products or procedures, patients may choose not to undergo our procedure, and physicians or others in the medical community may choose not to use our products. Furthermore, unsatisfactory patient outcomes or patient injury could cause negative publicity for our products, particularly in the early phases of product introduction.
We have limited experience in producing our core components and products, and if we are unable to manufacture our core components and products in high-quality commercial quantities successfully and consistently to meet demand, our growth will be limited.
We have experience manufacturing only limited quantities of rhCollagen, the recombinant human type I collagen used in our products. Our manufacturing capabilities will need to be further improved to meet the standard requirements for future clinical studies and for commercialization of our products. To manufacture our rhCollagen in quantities that we believe will be sufficient to produce our end products and meet anticipated market demand, we will need to increase manufacturing capacity, which will involve significant challenges. In addition, the development of commercial-scale, regulation-compliant manufacturing capabilities will require us to invest substantial additional funds and hire and retain the technical personnel who have the necessary manufacturing experience. We may not successfully complete any required increase to existing manufacturing processes in a timely manner, or at all. Our costs will be higher, and our challenges greater, if we decide to develop internal manufacturing capabilities to produce our end products.
9
If there is a disruption to our internal manufacturing operations, we will have no other means of production for the components and products from such operations until we restore the affected facilities or develop alternative manufacturing facilities, which would delay our clinical trials or cause us to be unable to meet commercial demand for our products. In such case, we may need to arrange for third-party manufacturing of our components and products, which would be expensive and time consuming, assuming we can identify an appropriate third party manufacturer. Additionally, any damage to or destruction of our facilities or equipment may significantly impair our ability to manufacture our components and products on a timely basis.
If we are unable to produce our products in sufficient quantities to meet anticipated customer demand, our revenues, business, and financial prospects would be harmed. The lack of experience we have in producing commercial quantities of our components and products may also result in quality issues and product recalls. Any product recall could be expensive and generate negative publicity, which could impair our ability to market our products and further affect our results of operations. Manufacturing delays related to quality control could negatively impact our ability to bring our technologies to market, harm our reputation, and decrease our revenues.
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell any of our products that obtain regulatory approval, we may be unable to generate any revenue.
We have limited experience in selling and marketing our products or any other products. To successfully commercialize our products we will need to develop these capabilities, either on our own or with others. We are seeking to enter into commercial alliances with third-party collaborators and distributors to utilize their marketing and distribution capabilities, but we may be unable to do so on favorable terms, if at all. If any future collaboration or distribution partners do not commit sufficient resources to commercialize our future products, and if we are unable to develop the necessary marketing capabilities on our own, we will be unable to generate sufficient product revenue to sustain our business. We will be competing with many companies that currently have extensive and well-funded marketing and sales operations. Without an internal team or the support of a third party to perform marketing and sales functions, we may be unable to compete successfully against these more established companies or successfully commercialize any of our products.
We face competition and rapid technological change and the possibility that our competitors may develop therapies or products that are more advanced or effective than ours, which could impair our ability to successfully commercialize our products.
We operate in the regenerative medicine field, which is rapidly changing. We have competitors both in the United States and internationally, including major multinational pharmaceutical companies, biotechnology companies, medical technology companies, and universities and other research institutions.
Many of our potential competitors have substantially greater financial, technical and other resources, such as larger research and development staff and experienced marketing and manufacturing organizations. Competition may increase further as a result of advances in the commercial applicability of technologies and greater availability of capital for investment in these industries. Our potential competitors may succeed in developing, acquiring, or licensing on an exclusive basis, products that are more effective or less costly than any products that we may develop, or achieve earlier patent protection, regulatory approval, product commercialization, and market penetration than us. Additionally, technologies developed by others may render our potential products uneconomical or obsolete, and we may not be successful in marketing our products against competitors.
We are not aware of any competitors that produce collagen from plants or that produce recombinant type I human collagen. However, our collagen-based products will compete with alternative solutions; for example, our VergenixSTR product will compete with companies that sell platelet-rich plasma, or PRP, kits. Our VergenixFG product will compete with companies that produce and market animal collagen-based products and collagen products produced from skin donations.
10
A variety of risks associated with international operations could harm our business.
If any of our products are approved for commercialization, it is our current intention to market them on a regional or worldwide basis in the jurisdictions where they may be approved, either alone or in collaboration with third parties. In addition, we may conduct development activities in various jurisdictions throughout the world. We expect that we will be subject to additional risks related to engaging in international operations, including:
| ● | different regulatory requirements for product approval in foreign countries; |
| ● | reduced protection for intellectual property rights; |
| ● | unexpected changes in tariffs, trade barriers, and regulatory requirements; |
| ● | economic weakness, including inflation, or political instability in particular foreign economies and markets; |
| ● | compliance with tax, employment, immigration, and labor laws for employees living or traveling abroad; |
| ● | foreign currency fluctuations, which could result in increased operating expenses and reduced revenue, and other obligations incident to doing business in another country; |
| ● | workforce uncertainty in countries where labor unrest is more common than in the United States and Israel; |
| ● | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and |
| ● | business interruptions resulting from geopolitical actions, including war and terrorism, or natural disasters including earthquakes, typhoons, floods, and fires. |
The insurance coverage and reimbursement status of newly approved products is uncertain. Failure to obtain or maintain adequate coverage and reimbursement for any of our products that are approved could limit our ability to market those products and compromise our ability to generate revenue.
The availability of reimbursement by governmental and private payors is essential for most patients to be able to afford expensive treatments. Sales of our products will depend substantially, both in Europe and in the United States, on the extent to which the costs of our products will be paid by health maintenance, managed care, pharmacy benefit, and similar healthcare management organizations, or reimbursed by government health administration authorities, private health coverage insurers, and other third-party payors. If reimbursement is not available, or is available only to limited levels, we may not be able to successfully commercialize our products. Even if we obtain coverage for our products, third-party payors may not establish adequate reimbursement amounts, which may reduce the demand for, or the price of, our products. If reimbursement is not available or is available only to limited levels, we may not be able to commercialize certain of our products.
Furthermore, publication of discounts by third-party payors or authorities may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unacceptable levels, we or our partner may elect not to commercialize our products in such countries, and our business and financial condition could be adversely affected.
11
Promotion of off-label uses of our products by physicians could adversely affect our business.
Any regulatory approval of our products is limited to those specific indications for which our products have been deemed safe and effective by the regulatory authorities. In addition, any new indication for an approved product also requires regulatory approval. If we produce an approved product, we will rely on physicians to use and administer it as we have directed and for the indications described on the labeling. It is not, however, uncommon for physicians to use in unapproved, or “off-label,” uses or in a manner that is inconsistent with the manufacturer’s directions. To the extent such off-label uses and departures from our administration directions become pervasive and produce results such as reduced efficacy or other adverse effects, the reputation of our products in the marketplace may suffer. In addition, off-label uses may cause a decline in our revenue or potential revenue, to the extent that there is a difference between the prices of our product for different indications.
Furthermore, while physicians may choose to use our products for off-label uses, our ability to promote the products is limited to those indications that are specifically approved by the regulators. Although regulatory authorities generally do not regulate the behavior of physicians, they do restrict communications by companies with respect to off-label use. If our promotional activities fail to comply with these regulations or guidelines, we may be subject to warnings from, or enforcement action by, these authorities. In addition, failure to follow regulation authorities’ rules and guidelines relating to promotion and advertising can result in the regulation authorities’ refusal to approve a product, the suspension or withdrawal of an approved product from the market, product recalls, fines, disgorgement of money, operating restrictions, injunctions, or criminal prosecution.
Risks Related to the Clinical Development and Regulatory Approval of Our Products
We currently depend heavily on the future success of our rhCollagen-based biological ink, or BioInk, VergenixSTR, and VergenixFG. Any failure to successfully develop, obtain regulatory approval for, and commercialize these products, independently or in cooperation with a third party collaborator, or the experience of significant delays in doing so, would compromise our ability to generate revenue and become profitable.
We have invested a significant portion of our efforts and financial resources in the development of BioInk, VergenixSTR, and VergenixFG. Our ability to generate product revenue from our products depends heavily on the successful development, approval, and commercialization of our products, which, in turn, depend on several factors, including the following:
| ● | our ability to continue and support our rhCollagen platform technology and programs; |
| ● |
our ability to establish and maintain strategic partnerships, including the United License Agreement; |
| ● | successfully completing our ongoing and future clinical trials and other studies required for our products; |
| ● | demonstrating and maintaining the safety and efficacy of our products at a sufficient level of statistical or clinical significance and otherwise obtaining marketing approvals from regulatory authorities; |
| ● | establishing successful sales and marketing arrangements for our products VergenixSTR and VergenixFG in the jurisdictions where they may be approved; |
| ● | the availability of coverage and reimbursement by healthcare payors for our products in the jurisdictions where they may be approved; |
| ● | establishing successful manufacturing arrangements with third-party manufacturers that are compliant with current good manufacturing practices, or cGMP, and which will ensure the development of a large scale manufacturing process and adequate facilities or being able to conduct such manufacturing ourselves; |
| ● | establishing a large scale facility as a second source for the manufacture of commercial quantities of our products, if approved; and |
| ● | other risks described in this “Risk Factors” section. |
12
Our products are based on novel technology, which makes it difficult to predict the time and cost of product development and potential regulatory approval.
We have concentrated our product research and development efforts on our novel rhCollagen technology. The FDA has approved very few plant-expressed products. We may experience development challenges in the future related to our technology, which could cause significant delays or unanticipated costs, and we may not be able to solve such development challenges. We may also experience delays in developing a sustainable, reproducible, and scalable manufacturing process or transferring that process to commercial partners, if we decide to do so, which may prevent us from completing our clinical trials or commercializing our products on a timely or profitable basis, if at all.
In addition, the clinical trial requirements of European regulatory authorities, the FDA, and other regulatory authorities and the criteria these regulators use to determine the safety and efficacy of a product vary substantially according to the type, complexity, novelty, and intended use and market of the potential products. The regulatory approval process for novel products such as ours can be more expensive and take longer than for other, better known or extensively studied products. Our products may also be designated by the FDA or other regulatory authorities as combination products, which include: (1) a product comprised of two or more regulated components, e.g., drug/device, biologic/device, drug/biologic, or drug/device/biologic, that are physically, chemically, or otherwise combined or mixed and produced as a single entity; (2) two or more separate products packaged together in a single package or as a unit and comprised of drug and device products, device and biological products, or biological and drug products; (3) a drug, device, or biological product packaged separately that according to its investigational plan or proposed labeling is intended for use only with an approved individually specified drug, device, or biological product where both are required to achieve the intended use, indication, or effect and where upon approval of the proposed product the labeling of the approved product would need to be changed, e.g., to reflect a change in intended use, dosage form, strength, route of administration, or significant change in dose; or (4) any investigational drug, device, or biological product packaged separately that according to its proposed labeling is for use only with another individually specified investigational drug, device, or biological product where both are required to achieve the intended use, indication, or effect. Combination Products containing a biologic/device then may be regulated as a biologic product, resulting in a longer regulatory approval process than the regulatory approval process for a medical device alone. Approvals by any regulatory authorities may not be indicative of what the FDA or other regulatory agencies may require for approval, and vice versa.
Regulatory requirements governing medical devices and other products for medical use have changed frequently and may continue to change in the future. Also, before a clinical trial can begin, an institutional review board, or IRB, at each institution at which a clinical trial will be performed must review the proposed clinical trial to assess the safety of the trial. In addition, adverse developments in clinical trials of comparable products conducted by others may cause European regulatory authorities, the FDA, or other regulatory authorities to change the requirements for approval of any of our products.
These regulatory agencies and additional or new requirements may lengthen the regulatory review process, require us to perform additional studies, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of our products, or lead to significant approval and post-approval limitations or restrictions. As we advance our products, we will be required to consult with these regulatory authorities, and comply with applicable requirements. If we fail to do so, we may be required to delay or discontinue development of our products. Delay or failure to obtain, or unexpected costs in obtaining, the regulatory approval necessary to bring a potential product to market could impair our ability to generate product revenue and to become profitable.
We may find it difficult to enroll patients in our clinical trials, and patients could discontinue their participation in our clinical trials, which could delay or prevent clinical trials of our products.
Identifying and qualifying patients to participate in clinical trials of our products is critical to our success. The timing of our clinical trials depends on our ability to recruit patients to participate in our clinical trials. We may experience delays in patient enrollment in the future. If patients are unwilling to participate in our clinical trials because of negative publicity from adverse events in the biotechnology, pharmaceutical or medical technology industries, or for other reasons, including competitive clinical trials for similar patient populations, the timeline for recruiting patients, conducting trials, and obtaining regulatory approval of potential products may be delayed. These delays could result in increased costs, delays in advancing our product development, delays in testing the effectiveness of our technology, or termination of the clinical trials altogether.
13
We may not be able to identify, recruit, and enroll a sufficient number of patients, or those with required or desired characteristics to achieve diversity in a trial, to complete our clinical trials in a timely manner. Patient enrollment is affected by factors including:
| ● | design of the trial protocol; |
| ● | size of the patient population; |
| ● | eligibility criteria for the trial in question; |
| ● | severity of the disease/wounds under investigation; |
| ● | perceived risks and anticipated benefits of the product under study; |
| ● | proximity and availability of clinical trial sites for prospective patients; |
| ● | availability of competing therapies, products, and clinical trials; |
| ● | efforts to facilitate timely enrollment in clinical trials; |
| ● | patient referral practices of physicians; and |
| ● | ability to monitor patients adequately during and after treatment. |
We are currently conducting clinical trials in Israel and intend to seek marketing approval in Europe, China and the United States. We may not be able to initiate or continue clinical trials if we cannot enroll a sufficient number of eligible patients to participate in the clinical trials required by European regulatory authorities, the FDA, or other regulatory authorities.
In addition, patients enrolled in our clinical trials may discontinue their participation at any time during the trial as a result of a number of factors, including withdrawing their consent or experiencing adverse clinical events, which may or may not be related to our products under evaluation. The discontinuation of patients in any one of our trials may cause us to delay or abandon such clinical trial, or cause the results from that trial not to be positive or sufficient to support a filing for regulatory approval of the applicable product.
Our clinical trials may not be successful or may be delayed.
Before obtaining marketing approval from regulatory authorities for the sale of our products or any future product, we must conduct clinical trials to demonstrate the safety in humans for European CE marking certification, and the safety and efficacy of our products in humans for other regulatory authorities such as China and the United States. From time to time, we work with contract research organizations, or CROs, which assist us in overseeing and implementing our clinical trials. Clinical trials are expensive, time consuming, and uncertain as to outcome. We cannot guarantee that any clinical trials will be conducted as planned or completed on schedule, if at all. We may not receive FDA regulatory approval for the conduct of any particular clinical trial in the United States or regulatory approval for conduct of such clinical trial in other countries. A failure of one or more clinical trials can occur at any stage of testing. Events that may prevent successful or timely completion of clinical development include:
| ● | delays in reaching a consensus with regulatory agencies on trial design; |
| ● | delays in reaching agreement on acceptable terms with prospective CROs and clinical trial sites; |
| ● | delays in obtaining required IRB approval at each clinical trial site; |
| ● | delays in recruiting suitable patients to participate in our clinical trials; |
14
| ● | imposition of a clinical hold by regulatory agencies, including after an inspection of our clinical trial operations or trial sites; |
| ● | failure by our CROs, other third parties or us to perform in accordance with clinical trial requirements or the FDA’s good clinical practices, or GCP, or applicable regulatory requirements in other countries; |
| ● | delays in the testing, validation, manufacturing, and delivery of our products to the clinical sites; |
| ● | delays in having patients complete participation in a trial or return for post-treatment follow-up; |
| ● | clinical trial sites or patients dropping out of a trial; |
| ● | occurrence of serious adverse events associated with the products that are viewed to outweigh their potential benefits; or |
| ● | changes in regulatory requirements and guidance that require amending or submitting new clinical trial protocols. |
Any inability to successfully complete preclinical and clinical development could result in additional costs to us or impair our ability to generate revenue from product sales. In addition, if we make manufacturing or design changes to our products, we may need to conduct additional studies to bridge our modified products to earlier versions. Clinical trial delays could also shorten any periods during which we may have the exclusive right to commercialize our products or allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our products.
If the results of our clinical trials are inconclusive or if there are safety concerns or adverse events associated with our products, we may:
| ● | fail to obtain, or be delayed in obtaining, marketing approval for our products; |
| ● | obtain approval for indications or patient populations that are not as broad as intended or desired; |
| ● | obtain approval with labeling that includes significant use or distribution restrictions or safety warnings; |
| ● | be required to perform additional clinical trials to support approval or be subject to additional post-marketing testing requirements; |
| ● | have regulatory authorities withdraw their approval of the product or impose restrictions on its distribution; |
| ● | be subject to the addition of labeling statements, such as warnings or contraindications; |
| ● | be sued; or |
| ● | experience damage to our reputation. |
Any of these events could prevent us from achieving or maintaining market acceptance of our products and impair our ability to commercialize our products.
15
Success in early clinical trials may not be indicative of results obtained in later trials.
There is a high failure rate for medical devices, drugs, and biologics proceeding through clinical trials. A number of companies in the pharmaceutical, biotechnology, and medical technology industries have suffered significant setbacks in later stage clinical trials even after achieving promising results in earlier stage clinical trials. Data obtained from preclinical and clinical activities are subject to varying interpretations, which may delay, limit, or prevent regulatory approval. In addition, regulatory delays or rejections may be encountered as a result of many factors, including the novelty of the product and changes in regulatory policy during the period of product development.
Even if we complete the necessary preclinical studies and clinical trials, we cannot predict when or if we will obtain regulatory approval to commercialize a product, or the approval may be for a more narrow indication than we expect.
We cannot commercialize a product until the appropriate regulatory authorities have reviewed and approved the product. Even if our products demonstrate safety and efficacy in clinical trials, the regulatory agencies may not complete their review processes in a timely manner, or we may not be able to obtain regulatory approval. Additional delays may result if an FDA Advisory Committee or other regulatory authority recommends non-approval or restrictions on approval. In addition, we may experience delays or rejections based upon additional government regulation from future legislation or administrative action, or changes in regulatory agency policy during the period of product development, clinical trials, and the review process. Regulatory agencies also may approve a treatment for fewer or more limited indications than requested or may grant approval subject to the performance of post-marketing studies. In addition, regulatory agencies may not approve the labeling claims that are necessary or desirable for the successful commercialization of our treatment.
Side effects may occur following treatment with our products which could make it more difficult for our products to receive regulatory approval.
Treatment with our products may cause side effects or other adverse events. In addition, since our products may in the future be administered in combination with other therapies, patients or clinical trial participants may experience side effects or other adverse events that are unrelated to our product, but may still impact the success of our clinical trials. Additionally, our products could potentially cause other adverse events that have not yet been predicted. The experience of side effects and adverse events in our clinical trials could make it more difficult to achieve regulatory approval of our products or, if approved, could negatively impact the market acceptance of such products.
Even if we obtain regulatory approval for a product, our products will remain subject to regulatory scrutiny.
Even if we obtain regulatory approval in a jurisdiction, the regulatory authority may still impose significant restrictions on the indicated uses or marketing of our products, or impose ongoing requirements for potentially costly post-approval studies or post-market surveillance. Advertising and promotional materials must comply with FDA, Federal Trade Commission, or FTC, and European and other countries’ regulatory requirements and are subject to review by the FDA, FTC or other governmental authorities, in addition to other potentially applicable federal and state laws.
The laws that may affect our operations in the United States include:
| ● | the federal Anti-Kickback Statute, which prohibits, among other things, persons from knowingly and willfully soliciting, receiving, offering, or paying remuneration, directly or indirectly, to induce, or in return for, the purchase or recommendation of an item or service reimbursable under a federal healthcare program, such as the Medicare and Medicaid programs; |
| ● | federal civil and criminal false claims laws and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other third-party payors that are false or fraudulent; |
| ● | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created new federal criminal statutes that prohibit executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters; |
| ● | HIPAA, as amended by the Health Information Technology and Clinical Health Act, or HITECH, and its implementing regulations, which imposes certain requirements relating to the privacy, security, and transmission of individually identifiable health information; |
16
| ● | the federal physician sunshine requirements under the Patient Protection and Affordable Care Act, which requires manufacturers of drugs, devices, biologics, and medical supplies to report annually to the Centers for Medicare and Medicaid Services, or CMS, information related to payments and other transfers of value to physicians, other healthcare providers, and teaching hospitals, and ownership and investment interests held by physicians and other healthcare providers and their immediate family members; and |
| ● | foreign and state law equivalents of each of the above federal laws, such as the U.S. Foreign Corrupt Practices Act, or the FCPA, and anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the applicable compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws that require manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures; and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways, thus complicating compliance efforts. |
The scope of these laws and our lack of experience in establishing the compliance programs necessary to comply with this complex and evolving regulatory environment increase the risks that we may violate the applicable laws and regulations.
In addition, product manufacturers and their facilities are subject to continual review and periodic inspections by the European regulatory authorities, the FDA, and other regulatory authorities for compliance with cGMP or any applicable European or other governmental regulations. If we or a regulatory agency discover previously unknown problems with a product such as adverse events of unanticipated severity or frequency or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions relative to that product or the manufacturing facility, including requiring recall or withdrawal of the product from the market or suspension of manufacturing.
If we fail to comply with applicable regulatory requirements following approval of any of our products, one or more regulatory authorities could:
| ● | issue a warning letter asserting that we are in violation of the law; |
| ● | seek an injunction or impose civil or criminal penalties or monetary fines; |
| ● | suspend or withdraw regulatory approval; |
| ● | suspend any ongoing clinical trials; |
| ● | seize our product; or |
| ● | refuse to allow us to enter into supply contracts, including government contracts. |
Any government investigation of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity and potentially lead to private litigation. The occurrence of any event or penalty described above may inhibit our ability to commercialize our products and generate revenues.
17
We have only limited experience in regulatory affairs and intend to rely on consultants and other third parties for regulatory matters, which may affect our ability or the time we require to obtain necessary regulatory approvals.
In January 2019, the U.S. Food and Drug Administration, or FDA, responded to the Company’s Pre-Request for Designation, or Pre-RFD, regarding product classification and jurisdictional assessment. The FDA’s Office of Combination Products, or OCP, determined that VergenixSTR should be classified as a combination product and should be assigned to the FDA’s Center for Biologics Evaluation and Research, or CBER. A Pre-RFD is FDA’s preliminary, nonbinding assessment of (1) the regulatory identity or classification of a product as a drug, device, biological product, or combination product, and (2) which FDA Center (i.e., CBER, Center for Drug Evaluation and Research, or CDER, or Center for Devices and Radiological Health, or CDRH,) will have primary jurisdiction for the premarket review and regulation of the product. Therefore, this classification and jurisdictional assessment is subject to change. We currently do not intend to pursue an FDA regulatory pathway to market for VergenixSTR in 2019. We have limited experience in preparing and filing the applications necessary to gain regulatory approvals for our products. Moreover, the products that are likely to result from our development programs are based on new technologies that have not been extensively used in humans. The regulatory requirements governing these types of products may be less well defined or more rigorous than for conventional products. As a result, we may experience a longer regulatory review process in connection with obtaining regulatory approvals, if any, of products that we develop. We intend to rely on independent consultants for regulatory services and compliance and product development and filings in Europe, the United States and elsewhere. Any failure by our consultants to properly advise us regarding, or properly perform tasks related to, regulatory submission and other requirements could compromise our ability to develop and obtain regulatory approval of our products.
We are subject to stringent regulation and any adverse regulatory action may materially adversely affect our financial condition and business operations.
Our products, development activities, and manufacturing processes are subject to extensive and rigorous regulation by numerous government agencies, including European regulatory authorities, the FDA, and other regulatory authorities. To varying degrees, each of these agencies monitors and enforces our compliance with laws and regulations governing the development, testing, manufacturing, labeling, marketing, and distribution of our products. The process of obtaining marketing approval or clearance in Europe, the United States, and other countries for new products or enhancements or modifications to existing products, could:
| ● | take a significant amount of time; |
| ● | require the expenditure of substantial resources; |
| ● | involve rigorous and expensive preclinical and clinical testing, as well as increased post-market surveillance; |
| ● | involve modifications, repairs, or replacements of our products; and |
| ● | result in limitations on the indicated uses of our products. |
We cannot be certain that we will receive required approval or clearance from European regulatory authorities, the FDA, or other regulatory authorities for new products or modifications to existing products on a timely basis. The failure to receive approval or clearance for significant new products or modifications to existing products on a timely basis could have a material adverse effect on our financial condition and results of operations.
Both before and after a product is commercially released, we have ongoing responsibilities under FDA regulations. For example, we are required to comply with the FDA’s Quality System Regulation, or QSR, which are the good manufacturing requirements that the FDA applies to medical devices, and which mandate that manufacturers adhere to certain requirements pertaining to, among other things, development of our products, validation of manufacturing processes, controls for purchasing product components, and documentation practices. As another example, FDA regulations require us to provide information to the FDA whenever there is evidence that reasonably suggests that a product may have caused or contributed to a death or serious injury, or that a malfunction occurred which would be likely to cause or contribute to a death or serious injury upon recurrence. Compliance with applicable regulatory requirements is subject to continual review and is monitored rigorously through, among other things, periodic inspections by the FDA, which may result in observations on Form 483 that require corrective action, and in some cases warning letters. If the FDA were to conclude that we are not in compliance with applicable laws or regulations, or that any of our products are ineffective or pose an unreasonable health risk, the FDA could ban such products, detain or seize such products, order a recall, repair, replacement, or refund of such products, or require us to notify health professionals and others that the devices present unreasonable risks of substantial harm to the public health.
18
The FDA has been increasing its scrutiny of the medical device, drugs, and biologics industries, and regulatory agencies are expected to continue to scrutinize the industry closely with inspections, with possible enforcement actions by the FDA or other agencies. Additionally, the FDA may restrict manufacturing and impose other operating restrictions, enjoin and restrain certain violations of applicable law pertaining to medical products, and assess civil or criminal penalties against our officers, employees, or us. The FDA may also recommend prosecution to the Department of Justice. Any adverse regulatory action, depending on its magnitude, may restrict us from effectively manufacturing, marketing, and selling our products. In addition, negative publicity and product liability claims resulting from any adverse regulatory action could have a material adverse effect on our financial condition and results of operations.
Finally, the FDA issued regulations regarding “Current Good Manufacturing Practice Requirements for Combination Products” on January 22, 2013. These regulations may apply to some of our products if they are designated by the FDA as combination products, which are products composed of two or more regulated components, such as a drug and a medical device. There have been and will be additional costs associated with compliance with the FDA Good Manufacturing Practice Requirements regulations for Combination Products.
Governmental regulations have become increasingly stringent and more common, and we may become subject to even more rigorous regulation by governmental authorities in various countries in the future. Penalties for a company’s non-compliance with governmental regulation could be severe, including revocation or suspension of a company’s business license and criminal sanctions.
The impact of healthcare reform and other changes in the healthcare industry and in healthcare spending is currently unknown, and may adversely affect our business model.
The commercial potential for our approved products, if any, could be affected by changes in healthcare spending and policy in Europe, in the United States, and in other countries. We operate in a highly regulated industry and new laws, regulations, or judicial decisions, or new interpretations of existing laws, regulations, or decisions, related to healthcare availability, the method of delivery, or payment for healthcare products and services could negatively impact our business, operations, and financial condition.
In addition to the level of commercial success of our products, if approved, our future prospects are also dependent on our ability to successfully develop a pipeline of additional products, and we may not be successful in our efforts in using our platform technologies to identify or discover additional products.
The success of our business depends primarily upon our ability to identify, develop, and commercialize products based on our platform technology. Although we have three products at various stages of development, our research programs may fail to identify other potential products for clinical development for a number of reasons. Our research methodology may be unsuccessful in identifying potential products or our potential products may be shown to have harmful side effects or may have other characteristics that may make the products unmarketable or unlikely to receive marketing approval.
If any of these events occur, we may be forced to abandon our development efforts for a program or programs. Research programs to identify new products require substantial technical, financial, and human resources. We may focus our efforts and resources on potential programs or products that ultimately prove to be unsuccessful.
19
Risks Related to Our Reliance on Third Parties
We may not be successful in establishing and maintaining strategic partnerships, which could adversely affect our ability to develop and commercialize our rhCollagen based BioInk, dermal fillers for aesthetics, VergenixSTR, and VergenixFG.
To successfully develop and commercialize our products, we will need substantial financial resources as well as expertise and physical resources and systems. We may elect to develop some or all of these physical resources and systems and expertise ourselves, or we may seek to collaborate with another company that can provide some or all of such physical resources and systems as well as financial resources and expertise. For example, in October 2018, CollPlant Ltd., or CollPlant, our wholly-owned subsidiary, entered into the United License Agreement, with Lung Biotechnology PBC, or LB, a public benefit corporation and wholly-owned subsidiary of United Therapeutics Corporation, pursuant to which CollPlant and LB will collaborate in 3D bio-printing development of engineered lungs or lung substitutes using our rhCollagen and BioInk.
We face significant competition in seeking appropriate partners for our products, and the negotiation process is time-consuming and complex. In order for us to successfully partner our products, potential partners must view our products as economically valuable in markets they determine to be attractive in light of the terms that we are seeking and other available products for licensing by other companies. Even if we are successful in our efforts to establish strategic partnerships, the terms that we agree upon may not be favorable to us, and we may not be able to maintain such strategic partnerships if, for example, development or approval of a product is delayed or sales of an approved product are disappointing. Any delay in entering into strategic partnership agreements related to our products could delay the development and commercialization of our products and reduce their competitiveness even if they reach the market. If we fail to establish and maintain strategic partnerships related to our products, we will bear all of the risk and costs related to the development and commercialization of our products, and we will need to seek additional financing, hire additional employees and otherwise develop expertise which we do not have and for which we have not budgeted.
The risks in a strategic partnership include the following:
| ● | the strategic partner may not apply the expected financial resources, efforts, or required expertise in developing the physical resources and systems necessary to successfully develop and commercialize a product; |
| ● | the strategic partner may not invest in the development of a sales and marketing force and the related infrastructure at levels that ensure that sales of the products reach their full potential; |
| ● | we may be required to undertake the expenditure of substantial operational, financial, and management resources; |
| ● | we may be required to issue equity securities that would dilute our existing shareholders’ percentage ownership; |
| ● | we may be required to assume substantial actual or contingent liabilities; |
| ● | we may not receive requisite regulatory approvals; |
| ● | strategic partners could decide to withdraw a development program, or move forward with a competing product developed either independently or in collaboration with others, including our competitors; |
| ● | disputes may arise between us and a strategic partner that delay the development or commercialization or adversely affect the sales or profitability of the product; or |
| ● | the strategic partner may independently develop, or develop with third parties, products that could compete with our products. |
In addition, a strategic partner for one or more of our products may have the right to terminate the collaboration at its discretion. For example, LB may terminate the United License Agreement at any time upon 30 days’ written notice with respect to the entirety of the United License Agreement and upon 30 days’ written notice with respect to its license and other rights under the United License Agreement relating to one or more CollPlant patents, on a patent-by-patent and country-by-country basis. Any early termination in a manner adverse to us could have a material adverse effect on our liquidity, financial condition and results of operations. Any termination may require us to seek a new strategic partner, which we may not be able to do on a timely basis, if at all, or require us to delay or scale back our development and commercialization efforts. The occurrence of any of these events could adversely affect the development and commercialization of our products and materially harm our business and stock price by delaying the development of our products, and the sale of any products that may be approved by the FDA or other regulatory agencies, by slowing the growth of such sales, by reducing the profitability of the product and/or by adversely affecting the reputation of the product.
20
Further, a strategic partner may breach an agreement with us, and we may not be able to adequately protect our rights under these agreements. Furthermore, a strategic partner will likely negotiate for certain rights to control decisions regarding the development and commercialization of our products, if approved, and may not conduct those activities in the same manner as we would do so.
We expect to depend upon third-party distributors and resellers for a significant portion of our sales.
We expect to rely primarily upon sales through independent distributors and resellers. While we are highly dependent upon acceptance of our products and solutions by such third parties and their active marketing and sales efforts relating to our products, most of our distributors and resellers will not be obligated to deal with us exclusively and are not contractually subject to minimum purchase requirements. In addition, some of our distributors and resellers may sell competing products or solutions. As a result, our distributors and resellers may give higher priority to products or services of our competitors, thereby reducing their efforts in selling our products and services.
There can be no assurance that such distributors and resellers will act as effective sales agents for us, that they will remain our partners, or that, if we terminate or lose any of them, we will be successful in replacing them. Any disruption in our distribution channels could adversely affect our business, operating results, and financial condition.
We expect to rely on third parties to conduct some or all aspects of our product manufacturing, protocol development, research, and preclinical and clinical testing, and these third parties may not perform satisfactorily.
We do not expect to independently conduct all aspects of our product manufacturing, protocol development, research, and preclinical and clinical testing. We currently rely, and expect to continue to rely, on third parties with respect to parts of these items.
Any of these third parties may terminate their engagements with us at any time or upon advance notice. If we need to enter into alternative arrangements, it could delay our product development activities. Our reliance on these third parties for research and development activities will reduce our control over these activities but will not relieve us of our responsibility to ensure compliance with all required regulations and study protocols.
If these third parties do not successfully carry out their contractual duties, meet expected deadlines, or conduct our studies in accordance with regulatory requirements or our stated study plans and protocols, we will not be able to complete, or may be delayed in completing, the preclinical studies and clinical trials required to support future FDA, European, or other approvals of our products.
Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured the products ourselves, including:
| ● | the inability to negotiate manufacturing agreements with third parties under commercially reasonable terms; |
| ● | reduced control as a result of using third-party manufacturers for all aspects of manufacturing activities; |
| ● | termination or non-renewal of manufacturing agreements with third parties in a manner or at a time that is costly or damaging to us; and |
| ● | disruptions to the operations of our third-party manufacturers or suppliers caused by conditions unrelated to our business or operations, including the bankruptcy of the manufacturer or supplier. |
Any of these events could lead to clinical trial delays or failure to obtain regulatory approval, or impact our ability to successfully commercialize future products. Some of these events could be the basis of action from European regulatory authorities, the FDA, or other regulatory authorities, including injunction, recall, seizure, or total or partial suspension of production.
21
If we or our third-party manufacturers on which we rely cannot manufacture our products at sufficient yields, we may experience delays in development, regulatory approval, and commercialization.
Completion of our clinical trials and commercialization of our products require access to, or development of, facilities to manufacture our products at sufficient yields and at a commercial scale. We have limited experience in large scale manufacturing, or managing third parties in manufacturing any of our products in the volumes that are expected to be necessary to support large-scale clinical trials and sales. Our efforts to establish these capabilities may not meet our requirements as to scale-up, yield, cost, potency, or quality in compliance with cGMP. Our clinical trials should be conducted with product produced under applicable cGMP regulations. Failure to comply with these regulations would delay the regulatory approval process. Even an experienced third-party manufacturer may encounter difficulties in production, including:
| ● | costs and challenges associated with scale-up and attaining sufficient manufacturing yields; |
| ● | supply chain issues, including the timely availability and shelf life requirements of raw materials and supplies; |
| ● | quality control and assurance; |
| ● | shortages of qualified personnel and capital required to manufacture large quantities of product; |
| ● | compliance with regulatory requirements that vary in each country where a product might be sold; |
| ● | capacity limitations and scheduling availability in contracted facilities; and |
| ● | natural disasters that affect facilities and possibly limit production. |
Any delay or interruption in the supply of our products could have a material adverse effect on our business and operations.
The regulatory authorities also may, at any time following approval of a product for sale, audit our manufacturing facilities or those of our third-party contractors. If any such inspection or audit identifies a failure to comply with applicable regulations or our product specifications or if a violation of applicable regulations, including a failure to comply with the product specifications, occurs independent of such an inspection or audit, we or the relevant regulatory authority may require remedial measures that may be costly or time consuming for us or a third party to implement and that may include the temporary or permanent suspension of a clinical trial or commercial sales or the temporary or permanent closure of a facility.
If we or any of our third-party manufacturers fail to maintain regulatory compliance, the FDA or the European authorities can impose regulatory sanctions including, among other things, refusal to approve a pending application for a new product or revocation of a pre-existing approval.
Additionally, if supply from one approved manufacturer is interrupted, there could be a significant disruption in commercial supply. Switching manufacturers may involve substantial costs and is likely to result in a delay in our desired clinical and commercial timelines.
These factors could cause the delay of clinical trials, regulatory submissions, required approvals, or commercialization of our products; cause us to incur higher costs; and prevent us from commercializing our products successfully. Furthermore, if our suppliers fail to meet contractual requirements, and we are unable to secure one or more replacement suppliers capable of production at a substantially equivalent cost, our clinical trials may be delayed or we could lose potential revenue.
22
We expect to rely on third parties to conduct, supervise, and monitor our clinical trials, and if these third parties perform in an unsatisfactory manner, it may harm our business.
We rely heavily on hospitals, clinic centers, and other institutions and third parties, including the principal investigators and their staff, to carry out our clinical trials in accordance with our clinical protocols and designs. We also rely on a number of CROs to assist in undertaking, managing, monitoring, and executing our ongoing clinical trials. We expect to continue to rely on CROs, clinical data management organizations, medical institutions, and clinical investigators to conduct our development efforts in the future. We compete with many other companies for the resources of these third parties, and large pharmaceutical and medical device companies often have significantly more extensive agreements and relationships with such third-party providers, and such third-party providers may prioritize the requirements of such large pharmaceutical and medical device companies over ours. The third parties on whom we rely may terminate their engagements with us at any time, which may cause delay in the development and commercialization of our products. If any such third party terminates its engagement with us or fails to perform as agreed, we may be required to enter into alternative arrangements, which would result in significant cost and delay to our product development program. Moreover, our agreements with such third parties generally do not provide assurances regarding employee turnover and availability, which may cause interruptions in the research on our products by such third parties.
Moreover, while our reliance on these third parties for certain development and management activities will reduce our control over these activities, it will not relieve us of our responsibilities. For example, European regulatory authorities, the FDA, and other regulatory authorities require compliance with regulations and standards, including GCP requirements, for designing, conducting, monitoring, recording, analyzing, and reporting the results of clinical trials to ensure that the data and results from trials are credible and accurate and that the rights, integrity, and confidentiality of trial participants are protected. Although we rely on third parties to conduct our clinical trials, we are responsible for ensuring that each of these clinical trials is conducted in accordance with its general investigational plan and protocol under legal and regulatory requirements. Regulatory authorities enforce these GCP requirements through periodic inspections of trial sponsors, principal investigators, and trial sites. If we or any of our CROs fail to comply with applicable GCP requirements, the clinical data generated in our clinical trials may be deemed unreliable, and European regulatory authorities, the FDA, or other regulatory authorities may require us to perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a regulatory authority, such regulatory authority will determine that any of our clinical trials comply with GCP requirements.
If CROs and other third parties do not successfully carry out their duties under their agreements with us, if the quality or accuracy of the data they obtain is compromised due to their failure to adhere to trial protocols or to regulatory requirements, or if they otherwise fail to comply with regulations and trial protocols or meet expected standards or deadlines, the trials of our products may not meet regulatory requirements. If trials do not meet regulatory requirements or if these third parties need to be replaced, the development of our products may be delayed, suspended, or terminated, or the results may not be acceptable. If any of these events occur, we may not be able to obtain regulatory approval of our products on a timely basis, at a reasonable cost, or at all.
Our reliance on third parties may require us to share our trade secrets, which increases the possibility that a competitor will discover them or that our trade secrets will be misappropriated or disclosed.
Because we rely on third parties to manufacture our products, and because we collaborate with various organizations and academic institutions on the advancement of our technology, we must, at times, share trade secrets with them. We seek to protect our proprietary technology in part by entering into confidentiality agreements and, if applicable, material transfer agreements, collaborative research agreements, consulting agreements, or other similar agreements with our strategic partners, advisors, employees, and consultants prior to beginning research or disclosing proprietary information. These agreements typically limit the rights of the third parties to use or disclose our confidential information, such as trade secrets. Despite these contractual provisions, the need to share trade secrets and other confidential information increases the risk that such trade secrets become known by potential competitors, are inadvertently incorporated into the technology of others, or are disclosed or used in violation of these agreements. Given that our proprietary position is based, in part, on our know-how and trade secrets, discovery by a third party of our trade secrets or other unauthorized use or disclosure would impair our intellectual property rights and protections in our products.
23
In addition, these agreements typically restrict the ability of our collaborators, advisors, employees, and consultants to publish data potentially relating to our trade secrets. Our academic collaborators typically have rights to publish data, provided that we are notified in advance and may delay publication for a specified time in order to secure our intellectual property rights arising from the collaboration. In other cases, publication rights are controlled exclusively by us, although in some cases we may share these rights with other parties. Despite our efforts to protect our trade secrets, our competitors may discover our trade secrets, either through breach of these agreements, independent development, or publication of information including our trade secrets in cases where we do not have proprietary or otherwise protected rights at the time of publication.
It could be difficult to replace some of our suppliers and equipment vendors.
Outside vendors provide key components, raw materials, and equipment used in the manufacture of our products. An uncorrected defect or supplier’s variation in a component or raw material, either unknown to us or incompatible with our manufacturing process, could harm our ability to manufacture products. We may not be able to find a sufficient alternative supplier in a reasonable time period, or on commercially reasonable terms, if at all, and our ability to produce and supply our products could be impaired.
If we were suddenly unable to purchase from one or more of these companies, we would need a significant period of time to qualify a replacement, and the production of any affected products could be disrupted. While it is our policy to maintain sufficient inventory of components so that our production will not be significantly disrupted even if a particular component or material is not available for a period of time, we remain at risk that we will not be able to qualify new components or materials quickly enough to prevent a disruption if one or more of our suppliers ceases production of important components or materials, or if we are unable to quickly procure replacement equipment.
Risks Related to Our Business Operations
Our future success depends on our ability to retain key employees, consultants, and advisors and to attract, retain, and motivate qualified personnel.
We are dependent on principal members of our executive team listed under “Management” in this Annual Report on Form 20-F, the loss of whose services may adversely impact the achievement of our objectives. While we have entered into employment agreements with each member of our senior management, any of them could leave our employment at any time, subject to advance notice periods. Recruiting and retaining other qualified employees, consultants, and advisors for our business, including scientific and technical personnel, will also be critical to our success. There is currently a shortage of skilled executives in our industry, which is likely to continue. As a result, competition for skilled personnel is intense and the turnover rate can be high. We may not be able to attract and retain personnel on acceptable terms given the competition among numerous pharmaceutical and medical device companies for individuals with similar skill sets. In addition, failure to succeed in clinical trials may make it more challenging to recruit and retain qualified personnel. The inability to recruit or loss of the services of any executive, key employee, consultant, or advisor may impede the progress of our research, development, and commercialization objectives.
Our collaborations with outside scientists and consultants may be subject to restriction and change.
We work with medical experts, chemists, biologists, and other scientists at academic and other institutions, and consultants who assist us in our research, development, and regulatory efforts, including the members of our scientific advisory board. In addition, these scientists and consultants have provided, and we expect that they will continue to provide, valuable advice regarding our programs and regulatory approval processes. These scientists and consultants are not our employees and may have other commitments that would limit their future availability to us. If a conflict of interest arises between their work for us and their work for another entity, we may lose their services. In addition, we are limited in our ability to prevent them from establishing competing businesses or developing competing products. For example, if a key scientist acting as a principal investigator in any of our clinical trials identifies a potential product that is more scientifically interesting to his or her professional interests, his or her availability to remain involved in our clinical trials could be restricted or eliminated.
24
Our business and operations would suffer in the event of computer system failures or security breaches.
Despite the implementation of security measures, our internal computer systems, and those of our contract research organizations, or CROs and other third parties on which we rely, are vulnerable to damage from computer viruses, unauthorized access, cyber-attacks, natural disasters, fire, terrorism, war, and telecommunication and electrical failures. If such an event were to occur and interrupt our operations, it could result in a material disruption of our drug development programs. For example, the loss of clinical trial data from ongoing or planned clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to our data or applications, loss of trade secrets or inappropriate disclosure of confidential or proprietary information, including protected health information or personal data of employees or former employees, access to our clinical data, or disruption of the manufacturing process, we could incur liability and the further development of our drug candidates could be delayed. We may also be vulnerable to cyber-attacks by hackers or other malfeasance. This type of breach of our cybersecurity may compromise our confidential information and/or our financial information and adversely affect our business or result in legal proceedings. Further, these cybersecurity breaches may inflict reputational harm upon us that may result in decreased market value and erode public trust.
We will need to expand our organization and we may experience difficulties in managing this growth, which could disrupt our operations.
As of March 15, 2019, we had 42 employees. As we mature and undertake the activities required to advance our products into later stage clinical development and to operate as a public company in the United States, we expect to expand our full-time employee base and to hire more consultants and contractors. Our management may need to divert a disproportionate amount of its attention away from our day-to-day activities and devote a substantial amount of time to managing these growth activities. We may not be able to effectively manage the expansion of our operations, which may result in weaknesses in our infrastructure, operational setbacks, loss of business opportunities, loss of employees, and reduced productivity among remaining employees. Our expected growth could require significant capital expenditures and may divert financial resources from other projects, such as the development of additional products. If our management is unable to effectively manage our growth, our expenses may increase more than expected, our ability to generate or grow revenue could be compromised, and we may not be able to implement our business strategy. Our future financial performance and our ability to commercialize products and compete effectively will depend, in part, on our ability to effectively manage any future growth.
Our employees, principal investigators, consultants, and commercial partners may engage in misconduct or other improper activities, including non-compliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of fraud or other misconduct by our employees, principal investigators, consultants, and commercial partners. Misconduct by these parties could include intentional failures to comply with regulations, provide accurate information to European regulatory authorities, the FDA and other regulatory authorities, comply with healthcare fraud and abuse laws and regulations, report financial information or data accurately, or disclose unauthorized activities to us. In particular, sales, marketing, and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing, and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs, and other business arrangements. Such misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and cause serious harm to our reputation. We have adopted a code of conduct applicable to all of our employees, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
25
We face potential product liability, and, if successful claims are brought against us, we may incur substantial liability and costs. If the use of our products harms patients, or is perceived to harm patients even when such harm is unrelated to our products, our regulatory approvals could be revoked or otherwise negatively impacted and we could be subject to costly and damaging product liability claims.
The use of our products in clinical trials and the sale of any products for which we obtain marketing approval exposes us to the risk of product liability claims. Product liability claims might be brought against us by consumers, healthcare providers, pharmaceutical and medical device companies, or others that sell or otherwise come into contact with our products. There is a risk that our products may induce adverse events. If we cannot successfully defend against product liability claims, we could incur substantial liability and costs. In addition, regardless of merit or eventual outcome, product liability claims may result in:
| ● | impairment of our business reputation; |
| ● | withdrawal of clinical trial participants; |
| ● | costs due to related litigation; |
| ● | distraction of management’s attention from our primary business; |
| ● | substantial monetary awards to patients or other claimants; |
| ● | the inability to commercialize our products; |
| ● | decreased demand for our products, if approved for commercial sale; and |
| ● | impairment of our ability to obtain product liability insurance coverage. |
We currently carry product liability insurance of $5.0 million for sales in Europe of VergenixFG and VergenixSTR. We intend to acquire product liability insurance before commercializing any of our other products. We believe our clinical trials liability insurance coverage is sufficient in light of our current clinical programs; however, we may not be able to obtain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to product liability. If we obtain marketing approval for any of our products, we intend to obtain insurance coverage to include the sale of commercial products, but we may not be able to obtain product liability insurance on commercially reasonable terms or in adequate amounts. On occasion, large judgments have been awarded in class action lawsuits based on medical treatments that had unanticipated adverse effects. A product liability claim or series of claims brought against us could cause our ADS or ordinary share price to decline and, if judgments exceed our insurance coverage, could materially and adversely affect our financial position.
Our development of rhCollagen relies upon the continued availability of tobacco plants, and any interruption in availability or supply of tobacco plants may delay production and adversely affect commercial utilization of our rhCollagen-based products.
Our products are all based on our recombinant human collagen extracted from tobacco plants. Any disruption to the supply of tobacco plants or any change in its availability for use would delay our production of collagen and adversely affect commercial utilization of our products.
The occurrence of severe adverse weather conditions, soil salination or crop diseases may have a potentially devastating impact upon our tobacco production. The effect of severe adverse weather conditions or the occurrence and effect of crop disease may reduce yields in our plants or require higher levels of investment to maintain yields, even when only a portion of the crop is damaged. We cannot assure you that severe future adverse weather conditions will not adversely impact our operating results and financial condition. Although some crop diseases are treatable, the cost of treatment is high, and we cannot assure that such events in the future will not adversely affect our operating results and financial condition.
26
If our existing rhCollagen production site or any new facility is damaged or destroyed, or production at this facility is otherwise interrupted, our business and prospects would be negatively affected.
We currently have a single, small-scale production site in Israel where we manufacture rhCollagen. Under the United License Agreement, LB is required to build a facility, or engage a manufacturer to build a facility, in the U.S. for the manufacture of our rhCollagen and BioInk in connection with the United License Agreement. There can be no assurance that such a facility will be built. If our existing production facility or the new facility, or the equipment in it, is damaged or destroyed, we likely would not be able to quickly or inexpensively replace our production capacity. Any new facility needed to replace our existing production facility would need to comply with the necessary regulatory requirements and be tailored to our production requirements and processes. We would need regulatory approval before using any products manufactured at a new facility in clinical trials or selling any products that are ultimately approved. Such an event could delay our clinical trials or, if any of our products are approved by the regulator, reduce or eliminate our product sales.
If we fail to comply with environmental, health, and safety laws and regulations, we could become subject to fines or penalties or incur costs that could have a material adverse impact on the success of our business.
We are subject to numerous environmental, health, and safety laws and regulations, including those governing laboratory procedures and the handling, use, storage, treatment, and disposal of hazardous materials and wastes. Our operations involve the use of hazardous materials, including chemicals and biological materials. Our operations also produce hazardous waste products. We generally contract with third parties for the disposal of these materials and wastes. We cannot eliminate the risk of contamination or injury from these materials. In the event of contamination or injury resulting from our use of hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of hazardous materials or other work-related injuries, this insurance may not provide adequate coverage against potential liabilities. In addition, we may incur substantial costs in order to comply with current or future environmental, health, and safety laws and regulations. These current or future laws and regulations may impair our research, development or production efforts. Failure to comply with these laws and regulations also may result in substantial fines, penalties, or other sanctions.
We may use our financial and human resources to pursue a particular research program or product and fail to capitalize on programs or products that may be more profitable or for which there is a greater likelihood of success.
Because we have limited resources, we may forego or delay pursuit of opportunities with certain programs or products or for indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on current and future research and development programs for products may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product, we may relinquish valuable rights to that product through strategic collaboration, licensing, or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product, or we may allocate internal resources to a product in a therapeutic area in which it would have been more advantageous to enter into a collaboration arrangement.
We are subject to foreign currency exchange risk, and fluctuations between the U.S. dollar and the NIS, the Euro, and other non-U.S. currencies may adversely affect our earnings and results of operations.
We currently operate in two different currencies. While the NIS is our functional and reporting currency and certain investments in our share capital have been denominated in NIS, our financial results may be adversely affected by fluctuations in currency exchange rates as a significant portion of our operating expenses, including development and manufacturing expenses, are denominated in U.S. dollars.
We are exposed to the risks that the U.S. dollar may appreciate relative to the NIS, In such event, the dollar-denominated results of operations would be adversely affected. We cannot predict any future trends in the rate of inflation in Israel or the rate of devaluation (if any) of the NIS against the dollar. For example, the average exchange rate of the dollar against the NIS increased in 2018 and decreased in 2017 and 2016. Market volatility and currency fluctuations may limit our ability to cost-effectively hedge against our foreign currency exposure. Hedging strategies may not eliminate our exposure to foreign exchange rate fluctuations and may involve costs and risks of their own, such as devotion of management time, external costs to implement the strategies, and potential accounting implications. Foreign currency fluctuations, independent of the performance of our underlying business, could lead to materially adverse results or could lead to positive results that are not repeated in future periods.
27
Risks Related to Our Intellectual Property
We have an extensive worldwide patent portfolio. The cost of maintaining our patent protection is high and maintaining our patent protection requires continuous review and compliance in order to maintain worldwide patent protection. We may not be able to effectively maintain our intellectual property position throughout the major markets of the world.
The U.S. Patent and Trademark Office, or U.S. PTO, and foreign patent authorities require maintenance fees and payments as well as continued compliance with a number of procedural and documentary requirements. Non-compliance may result in abandonment or lapse of the subject patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Non-compliance may result in reduced royalty payments for lack of patent coverage in a particular jurisdiction from our collaboration partners or may result in competition, either of which could have a material adverse effect on our business.
We have made, and will continue to make, certain strategic decisions in balancing costs and the potential protection afforded by the patent laws of certain countries. As a result, we may not be able to prevent third parties from practicing our inventions in all countries throughout the world, or from selling or importing products made using our inventions in and into the United States or other countries. Third parties may use our technologies in territories in which we have not obtained patent protection to develop their own products and, further, may infringe our patents in territories which provide inadequate enforcement mechanisms, even if we have patent protection. Such third-party products may compete with our products, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
If we are unable to obtain or protect intellectual property rights related to our products, we may not be able to obtain exclusivity for our products or prevent others from developing similar competitive products.
We rely upon a combination of granted patents, pending patent applications, trade secret protection, and confidentiality agreements to protect the intellectual property related to our products. The strength of patents in the field of regenerative medicine involves complex legal and scientific questions and can be uncertain. The patent applications that we own may fail to result in issued patents with claims that cover our products in the United States or in other countries. There is no assurance that all of the potentially relevant prior art relating to our patents and patent applications has been found, which can invalidate a patent or prevent a patent from issuing from a pending patent application. Even if patents do successfully issue and even if such patents cover our products, third parties may challenge their validity, enforceability, or scope, which may result in the patent claims being narrowed or invalidated. Furthermore, even if they are unchallenged, our patents and patent applications may not adequately protect our intellectual property, provide exclusivity for our products, or prevent others from designing around our claims. Any of these outcomes could impair our ability to prevent competition from third parties.
Our ability to attract third parties to collaborate with us to develop products and our ability to commercialize future products may be adversely affected if the patent applications we hold with respect to our techniques or products fail to issue, if the breadth or strength of our patent protection is threatened, or if our patent portfolio fails to provide meaningful exclusivity for our products. Third parties may challenge their validity or enforceability of our patents or patents that issue in the future from our patent applications, which may result in such patents being narrowed, invalidated, or held unenforceable. Even if our patents and patent applications are not challenged by third parties, they may not prevent others from designing around our claims and may not otherwise adequately protect our products. If the breadth or strength of protection provided by the patents and patent applications we hold with respect to our products is threatened, our ability to commercialize our products may be adversely effected.
Discoveries are generally published in the scientific literature well after their actual development, and patent applications in the United States and other countries are typically not published until 18 months after filing and in some cases are never published. Therefore, we cannot be certain that we were the first to make the inventions claimed in our owned granted patents or patent applications, or that we were the first to file for patent protection covering such inventions. Subject to meeting other requirements for patentability, for United States patent applications filed prior to March 16, 2013, the first to invent the claimed invention is entitled to receive patent protection for that invention while, outside the United States, the first to file a patent application encompassing the invention is entitled to patent protection for the invention. In addition, patents have a limited lifespan. In the United States, the expiration of a patent is generally 20 years from the earliest non-provisional filing date. Various extensions may be available, but the life of a patent, and the protection it affords, is limited. Once the patent life has expired for a product, we may be open to competition from third party products, including products that are copies of our products. This risk is material in light of the length of the development process of our products and lifespan of our current patent portfolio.
28
In addition to the protection afforded by patents, we rely on trade secret protection and confidentiality agreements to protect our proprietary know-how and other proprietary information that is not patentable or that we elect not to patent. For example, many of our discovery, development, and manufacturing processes involve proprietary know-how, information, or technology that is not covered by patents. We seek to protect our trade secrets and proprietary technology and processes, in part, by entering into confidentiality agreements with our employees, consultants, scientific advisors, and contractors. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. Security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. Although we expect all of our employees and consultants to assign their inventions to us, and all of our employees, consultants, advisors, and any third parties who have access to our proprietary know-how, information, or technology to enter into confidentiality agreements, we cannot provide any assurances that all such agreements have been duly executed, that our trade secrets and other confidential proprietary information will not be disclosed, or that competitors will not otherwise gain access to our trade secrets or independently develop substantially equivalent information and techniques. Misappropriation or unauthorized disclosure of our trade secrets could impair our competitive position and may have a material adverse effect on our business. Additionally, if the steps taken to maintain our trade secrets are deemed inadequate, we may have insufficient recourse against third parties for misappropriating the trade secret. In addition, others may independently discover our trade secrets and proprietary information. For example, the FDA, as part of its Transparency Initiative, is currently considering whether to make additional information publicly available on a routine basis, including information that we may consider to be trade secrets or other proprietary information, and it is not clear at the present time how the FDA’s disclosure policies may change in the future, if at all.
Further, the laws of some countries do not protect proprietary rights to the same extent or in the same manner as the laws of the United States. As a result, we may encounter significant problems in protecting and defending our intellectual property both in the United States and in other countries. If we are unable to prevent material disclosure of the non-patented intellectual property related to our technologies to third parties, and there is no guarantee that we will have any such enforceable trade secret protection, we may not be able to establish or maintain a competitive advantage in our market.
Third-party claims of intellectual property infringement may prevent or delay our development and commercialization efforts.
Our commercial success depends in part on our avoiding infringement of the patents and proprietary rights of third parties. There is a substantial amount of litigation, both within and outside the United States, involving patents and other intellectual property rights in the biotechnology and pharmaceutical industries, including patent infringement lawsuits, interferences, oppositions, and inter partes review proceedings before the U.S. PTO, and corresponding foreign patent offices. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we are pursuing development technologies. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our products may be subject to claims of infringement of the patent rights of third parties.
Third parties may assert that we are employing their proprietary technology without authorization. There may be third-party patents or patent applications with claims to materials, formulations, methods of manufacture, or methods for treatment related to the use or manufacture of our products. Because patent applications can take many years to issue, there may be currently pending patent applications which may later result in issued patents that our products may be accused of infringing. In addition, third parties may obtain patents in the future and claim that use of our technologies infringes upon these patents. If any third-party patents were held by a court of competent jurisdiction to cover the manufacturing process of any of our products or any final product itself, the holders of any such patents may be able to block our ability to commercialize such product unless we obtained a license under the applicable patents, or until such patents expire. Similarly, if any third-party patents were held by a court of competent jurisdiction to cover aspects of our formulations, processes for manufacture, or methods of use, the holders of any such patents may be able to block our ability to develop and commercialize the applicable product unless we obtained a license or until such patent expires. In either case, such a license may not be available on commercially reasonable terms or at all.
29
The patent landscape in competitive product areas is highly complex and there may be patents of third parties of which we are unaware that may result in claims of infringement. Accordingly, there can be no assurance that our products do not infringe proprietary rights of third parties. Parties making claims against us may obtain injunctive or other equitable relief, which could effectively block our ability to further develop and commercialize one or more of our products. Defense of such claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of financial and employee resources from our business. In the event of a successful claim of infringement against us, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, pay royalties, redesign our infringing products, or obtain one or more licenses from third parties, which may be impossible or require substantial time and monetary expenditure.
We intend, if necessary, to vigorously enforce our intellectual property in order to protect the proprietary position of our products. Active efforts to enforce our patents may include litigation, post-grant patent challenges, administrative proceedings, or all of the foregoing, depending on the potential benefits that might be available from those actions and the costs associated with undertaking those efforts against third parties. We review and monitor publicly available information regarding products that may be competitive with our products and intend to assert our intellectual property rights where appropriate.
We may enter into license agreements with third parties, and if we fail to comply with our obligations in such agreements under which we license intellectual property rights from third parties or otherwise experience disruptions to our business relationships with our licensors, we could lose license rights that are important to our business.
We may need to obtain licenses from third parties to advance our research or allow commercialization of our products. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all. In that event, we may be required to expend significant time and resources to develop or license replacement technology. If we are unable to do so, we may be unable to develop or commercialize the affected products.
We may be involved in lawsuits or administrative proceedings to obtain, protect or enforce our patents, which could be expensive, time consuming, and unsuccessful.
Competitors may infringe our patents. To counter infringement or unauthorized use, we may be required to file an infringement suit, which can be expensive and time consuming. In addition, in an infringement proceeding, the defendant may file a countersuit, challenging the validity or enforceability of our patent. In that case, a court may decide that a patent of ours is not valid, is unenforceable, or is not infringed, or it may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
We may not be able to prevent misappropriation of our intellectual property rights, particularly in countries where the laws may not protect those rights.
We may be involved in interference proceedings in the U.S. PTO that are provoked by third parties or provoked by us when there appears to be the same subject matter claimed in our patents or patent applications and the third parties’ patents or patent applications, in order to determine the priority of inventions. An unfavorable outcome could require us to cease using the related technology, to lose our patent claims partially or in entirety, or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Our defense of interference proceedings may fail and, even if successful, may result in substantial costs and distract our management and other employees.
30
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. There could also be public announcements of the results of hearings, motions, or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a material adverse effect on the trading price of our ordinary shares or ADSs.
Recent patent reform legislation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents.
On September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to U.S. patent law, including provisions that affect the way patent applications are prosecuted and also affect patent litigation. The U.S. PTO has developed regulations and procedures to govern administration of the Leahy-Smith Act, and many of the substantive changes to patent law associated with the Leahy-Smith Act, and in particular, the first to file provisions which were enacted March 16, 2013. However, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. The Leahy-Smith Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents. We may become involved in post-grant proceedings challenging our patents or the patents of others, and the outcome of any such proceedings are highly uncertain. An unfavorable outcome in any such proceedings could reduce the scope of, or invalidate, our patent rights, allow third parties to commercialize our technology and compete directly with us, or result in our inability to manufacture, develop, or commercialize our products without infringing the patent rights of others.
We may be subject to claims that our employees, consultants, or independent contractors have wrongfully used or disclosed confidential information of third parties or, that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
Certain of our employees and personnel were previously employed at universities, medical institutions, or other biotechnology or pharmaceutical companies. Although we try to ensure that our employees, consultants, and independent contractors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or our employees, consultants, or independent contractors have inadvertently or otherwise used or disclosed intellectual property, including trade secrets or other proprietary information, of any of our employee’s former employer or other third parties. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees. Furthermore, universities or medical institutions who employ some of our key employees and personnel in parallel to their engagement by us may claim that intellectual property developed by such person is owned by the respective academic or medical institution under the respective institution, intellectual property policy or applicable law.
We may become subject to claims for remuneration or royalties for assigned service invention rights by our employees, which could result in litigation and adversely affect our business.
A significant portion of our intellectual property has been developed by our employees in the course of their employment for us. Section 134 of the Israeli Patents Law, 5727-1967, or the Patents Law, grants employees the right to receive consideration for service inventions unless otherwise provided in an agreement between the parties. According to a decision by the special Committee for Compensations and Royalties formed under the Patents Law, or the Committee, an employee’s right to receive consideration for service inventions is a personal right and is entirely separate from the proprietary rights in such invention. A decision in May 2014 by the Committee clarifies that the right to receive consideration under Section 134 can be waived and that such waiver does not necessarily have to be explicit. However, the Committee has the authority to examine, on a case by case basis, the general contractual framework between the parties, using interpretation rules of the general Israeli contract laws. Although such decision seems to alleviate the requirement to obtain an explicit waiver for royalties for service inventions under Section 134 of the Patents Law, to the extent that there is no explicit waiver in an employment agreement, the existence of such waiver will be subject to the interpretation of the Committee. Further, the Committee has not yet determined one specific formula for calculating this remuneration (but rather uses the criteria specified in the Patents Law) nor the criteria or circumstances under which an employee’s waiver of his right to remuneration will be disregarded. We generally enter into assignment-of-invention agreements with our employees pursuant to which such individuals assign to us all rights to any inventions created in the scope of their employment or engagement with us. Although our employees have agreed to assign to us service invention rights, we may face claims demanding remuneration in consideration for assigned inventions. As a consequence of such claims, we could be required to pay additional remuneration or royalties to our current or former employees, or be forced to litigate such claims, which could negatively affect our business.
31
We may be subject to claims challenging the inventorship or ownership of our patents and other intellectual property.
We may be subject to claims that former employees, collaborators, or other third parties have an ownership interest in our patents or other intellectual property. Ownership disputes may arise in the future, for example, from conflicting obligations of consultants or others who are involved in developing our products. Litigation may be necessary to defend against these and other claims challenging inventorship or ownership. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, valuable intellectual property. Such an outcome could have a material adverse effect on our business. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees.
Obtaining and maintaining our patent protection requires compliance with various procedural, document submissions, fee payments, and other requirements imposed by governmental patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees, and various other governmental fees on patents and applications are and will be due to be paid to the U.S. PTO and various governmental patent agencies outside of the United States in several stages over the lifetime of the patents and applications. The U.S. PTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment, and other similar provisions during the patent application process. There are situations in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction.
Issued patents covering our products could be found invalid or unenforceable if challenged in court or in administrative proceedings.
If we initiate legal proceedings against a third party to enforce a patent covering one of our products, the defendant may contend that the patent covering our product is invalid, unenforceable, or fails to cover the product or the infringing product. In patent litigation in the United States, defendants commonly allege that asserted patent claims are invalid and unenforceable. Grounds for a validity challenge could be an alleged failure to meet one or more of several statutory requirements, including lack of novelty, obviousness, lack of written description, indefiniteness, and non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the U.S. PTO, or made a misleading statement, during prosecution. Third parties may also raise similar claims before administrative bodies in the United States or abroad, even outside the context of litigation. Such mechanisms include re-examination, post grant review, and equivalent proceedings in foreign jurisdictions, such as opposition proceedings. Such proceedings could result in revocation, amendments to our patent claims, or statements being made on the record such that our claims may no longer be construed to cover our products. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we and the patent examiner were unaware during prosecution. If a defendant were to prevail on a legal assertion of invalidity, unenforceability, or non-infringement, we would lose at least part, and perhaps all, of the patent protection on our products. For example, as further described below, in July 2017, Fibrogen, Inc., or Fibrogen, prevailed in an administrative challenge to one of our patents in Europe, resulting in the revocation of the patent and the abandonment of another patent. Even if resolved in our favor, litigation, or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses, and could distract our technical and management personnel from their normal responsibilities. Moreover, third parties may continue to initiate new proceedings in the United States and foreign jurisdictions to challenge our patents from time to time.
32
In addition, there could be public announcements of the results of hearings, motions, or other interim proceedings or developments, and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the market price of our ordinary shares or ADSs. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing, or distribution activities.
Three issued patents in Europe covering our product were administratively challenged by Fibrogen.
Our European Patent No. 1 809 751 entitled “Collagen Producing Plants and Methods of Generating and Using Same,” was granted by the European Patent Office, or the EPO, on September 1, 2010. On June 1, 2011, Fibrogen initiated an opposition proceeding with the EPO, seeking revocation of the patent in its entirety on the grounds that the claims were not supported by the contents of the patent, were not novel, and were not inventive. On January 22, 2013, the EPO issued its decision to maintain the patent in amended form with claims that cover genetically modified plants that produce collagen.
On June 3, 2013, Fibrogen, Inc. appealed the decision. On August 1, 2013, we filed an appeal, seeking to expand the scope of the patent. In July 2017, the EPO revoked the patent.
Our European Patent No. 2 357 241 entitled “Collagen Producing Plants and Methods of Generating and Using Same,” a divisional of European Patent No. 1 809 751, was granted by the EPO, on March 4, 2015. On December 10, 2015, Fibrogen initiated an opposition proceeding with the EPO, seeking revocation of the patent in its entirety on the grounds that the claims were not supported by the contents of the patent, were not novel, and were not inventive. On August 16, 2016, we filed a response thereto. In September 2017, we abandoned the patent and consequently the patent was revoked by the EPO in February 2018.
Our European Patent No. EP2816117 entitled “Collagen Producing Plants and Methods of Generating and Using Same,” a divisional of European Patent No. 1 809 751, was granted by the EPO, on November 30, 2016. On August 30, 2017, Fibrogen initiated an opposition proceeding with the EPO, seeking revocation of the patent in its entirety on the grounds that the claims were not supported by the contents of the patent, and were not inventive. Due to our decision not to respond in the opposition proceedings, the patent was revoked in July 2018.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
As is the case with other companies in our industry, our success is heavily dependent on intellectual property, particularly patents. Obtaining and enforcing patents in the biotechnology industry involve both technological and legal complexity, and therefore is costly, time consuming, and inherently uncertain. In addition, in recent years, the United States enacted and implemented wide-ranging patent reform legislation. Recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in some situations. In addition to increasing uncertainty with regard to our ability to obtain patents in the future, this combination of events has created uncertainty with respect to the value of patents that had already been granted. The patent laws and regulations may change in unpredictable ways through actions of the U.S. Congress, the federal courts, and the U.S. PTO, in the future, and any changes may adversely affect our ability to obtain new patents or to enforce our existing patents and patents that we might obtain in the future.
33
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting, and defending patents on products in all countries throughout the world would be prohibitively expensive, and our intellectual property rights in some countries outside the United States can be less extensive than those in the United States. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as federal and state laws in the United States. Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the United States, or from selling or importing products made using our inventions in and into the United States or other jurisdictions. Potential competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products and may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as in the United States. These products may compete with our products, if approved, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets, and other intellectual property protection, particularly those relating to biotechnology products, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our proprietary rights generally. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly and our patent applications at risk of not issuing, and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
Intellectual property rights do not address all potential threats to any competitive advantage we may have.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations, and intellectual property rights may not adequately protect our business or permit us to maintain our competitive advantage. The following examples are illustrative:
| ● | Others may be able to make products that are the same as or similar to our current or future products but that are not covered by the claims of the patents that we own or have exclusively licensed. |
| ● | We or any of our licensors or strategic partners might not have been the first to make the inventions covered by the issued patent or pending patent application that we own or have exclusively licensed. |
| ● | We or any of our licensors or strategic partners might not have been the first to file patent applications covering certain of our inventions. |
| ● | Others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our intellectual property rights. |
| ● | The prosecution of our pending patent applications may not result in granted patents. |
| ● | Granted patents that we own or have exclusively licensed may not provide us with any competitive advantages, or may be held invalid or unenforceable, as a result of legal challenges by our competitors. |
| ● | Patent protection on our products may expire before we are able to develop and commercialize the product, or before we are able to recover our investment in the product. |
| ● | Our competitors might conduct research and development activities in the United States and other countries that provide a safe harbor from patent infringement claims for such activities, as well as in countries in which we do not have patent rights, and may then use the information learned from such activities to develop competitive products for sale in markets where we intend to market our products. |
34
Risks Related to the Offering and Ownership of the ADSs
The market price of the ADSs may be highly volatile.
Since our listing of the ADSs on the Nasdaq Capital Market on January 31, 2018 and which were previously quoted on the OTC from March 2015 to January 30, 2018, an active market has not developed. You may not be able to sell your ADSs quickly or at the market price if trading in the ADSs is not active.
The market price of the ADSs is likely to be volatile. Our ADS price could be subject to wide fluctuations in response to a variety of factors, including the following:
| ● | adverse results or delays in preclinical studies or clinical trials; |
| ● | reports of adverse events in other similar products or clinical trials of such products; |
| ● | inability to obtain additional funding; |
| ● | any delay in filing a regulatory submission for any of our products and any adverse development or perceived adverse development with respect to the FDA’s review or European authorities’ review of that regulatory submission; |
| ● | failure to develop successfully and commercialize our products and future products; |
| ● | failure to enter into or maintain strategic collaborations; |
| ● | failure by us or strategic collaboration partners to prosecute, maintain, or enforce our intellectual property rights; |
| ● | changes in laws or regulations applicable to future products; |
| ● | inability to scale up our manufacturing capabilities (including in Israel), inability to obtain adequate product supply for our products, or the inability to do so at acceptable prices; |
| ● | adverse regulatory decisions, including by the IIA under the Innovation Law; |
| ● | introduction of new products, services, or technologies by our competitors; |
| ● | failure to meet or exceed financial projections we may provide to the public; |
| ● | failure to meet or exceed the financial expectations of the investment community; |
| ● | the perception of the biotechnology industry by the public, legislatures, regulators, and the investment community; |
| ● | announcements of significant acquisitions, strategic partnerships, joint ventures, or capital commitments by us or our competitors; |
| ● | disputes or other developments relating to proprietary rights, including patents, litigation matters, and our ability to obtain patent protection for our technologies; |
| ● | additions or departures of key scientific or management personnel; |
| ● | significant lawsuits, including patent or shareholder litigation; |
| ● | changes in the market valuations of similar companies; |
| ● | sales of our ordinary shares or ADSs by us or our shareholders in the future; and |
| ● | trading volumes of our ordinary shares and ADSs. |
35
In addition, companies trading in the stock market in general, and life science companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our ordinary shares, regardless of our actual operating performance.
We may not be able to maintain our listing on the Nasdaq Capital Market.
Nasdaq has established certain standards for the continued listing of a security on the Nasdaq Capital Market. The standards for continued listing include, among other things, that the minimum bid price for the listed securities not fall below $1.00 per share for a period of 30 consecutive trading days and that we maintain a minimum of $2,500,000 in shareholders’ equity. In the future we may not satisfy the Nasdaq’s continued listing standards. If we do not satisfy any of the Nasdaq’s continued listing standards, our ADSs could be delisted. Any such delisting could adversely affect the market liquidity of our ADSs and the market price of our ADSs could decrease. A delisting could adversely affect our ability to obtain financing for our operations or result in a loss of confidence by investors, customers, suppliers or employees.
We incur significant additional costs as a result of being a public company subject to SEC reporting requirements in the United States, and our management is required to devote substantial additional time to new compliance initiatives as well as to compliance with ongoing United States reporting requirements.
As a U.S. public reporting company, we are incurring significant additional accounting, legal, and other expenses in the future. Our management and other personnel need to devote substantial time to the compliance requirements of being a U.S. public company; in addition, the implementation of such compliance processes and systems may require us to hire outside consultants and incur other significant costs. Any future changes in the laws and regulations affecting public companies in the United States and the rules and regulations adopted by the SEC and the Nasdaq Capital Market, for so long as they apply to us, will result in increased costs to us as we respond to such changes. These laws, rules, and regulations could make it more difficult or more costly for us to obtain certain types of insurance, including director and officer liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these requirements could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, on our board committees, if any, or as senior management.
Our principal shareholders, management and directors beneficially own a significant percentage of our ordinary shares and will be able to exert significant influence over matters subject to shareholder approval.
As of March 15, 2019, our senior management, directors, and five percent or more shareholders and their affiliates beneficially owned approximately 57% of our ordinary shares. These shareholders will be able to significantly influence all matters requiring shareholder approval, except for decisions that require a special majority at a shareholders’ meeting. For example, these shareholders, if they were to act together, may be able to significantly influence elections of directors (other than our external directors, within the meaning of Israeli law, as described under “Management—External Directors”), amendments of our organizational documents, or approval of any merger, sale of assets, or other major corporate transaction. This may prevent or discourage unsolicited acquisition proposals or offers for our ordinary shares that you may believe are in your best interest as one of our shareholders.
In addition, but for the application of blocker provisions in certain security instruments that subject the conversion or exercise of such instruments, as applicable, to 4.99% beneficial ownership limitations and the need to conduct a special tender offer if Alpha wishes to hold 25% or more of the voting rights in the Company, Alpha would beneficially own approximately 35% of our outstanding ordinary shares as of March 15, 2019. If Alpha were to convert or exercise such instruments to the maximum extent possible, absent such blocker provisions and special tender offer requirement, Alpha would have the ability to also obtain a substantial interest in our Company. This concentration and potential further concentration of ownership could have the effect of delaying a change in our control or otherwise discouraging a potential acquirer from attempting to obtain control of us, which could in turn have an adverse effect on the market price of our ordinary shares or ADSs or prevent our shareholders from realizing a premium over the then-prevailing market price for their ordinary shares or ADSs. Furthermore, because of the large number of shares that may be issued from time to time under security instruments issued to Alpha, Meitav Dash and Ami Sagy, there may be an adverse effect on the market because of the quantity and regularity of conversion and/or exercise and sale of those shares, or even the potential of those shares being sold. Therefore, there may be limited demand and excessive price and volume volatility.
36
If we fail to maintain an effective system of internal control over financial reporting, we may not be able to accurately report our financial results or prevent fraud. As a result, our shareholders could lose confidence in our financial and other public reporting, which would harm our business and the trading price of the ADSs.
Effective internal controls over financial reporting are necessary for us to provide reliable financial reports. Together with adequate disclosure controls and procedures, effective internal controls are designed to prevent fraud. Any failure to implement required new or improved controls or difficulties encountered in their implementation could cause us to fail to meet our reporting obligations. In addition, any testing by us conducted in connection with Section 404 of the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses, may require prospective or retroactive changes to our financial statements, or may identify other areas for further attention or improvement. Inferior internal controls could also cause investors to lose confidence in our reported financial information, which could have a negative effect on the trading price of the ADSs.
We are required to disclose changes made in our internal controls and procedures on an annual basis and our management is required to assess the effectiveness of these controls annually. However, for as long as we are an “emerging growth company” under the Jumpstart Our Business Startups Act, or the JOBS Act, our independent registered public accounting firm will not be required to attest to the effectiveness of our internal controls over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act. We could be an emerging growth company for up to five years from the date of the first sale of our ADSs pursuant to an effective registration statement. An independent assessment of the effectiveness of our internal controls could detect problems that our management’s assessment might not. Undetected material weaknesses in our internal controls could lead to financial statement restatements and require us to incur the expense of remediation.
We are an “emerging growth company” and a “foreign private issuer,” and we cannot be certain if the reduced reporting requirements applicable to emerging growth companies and foreign private issuers will make the ADSs less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act. For as long as we continue to be an emerging growth company, we may take advantage of exemptions from various reporting requirements that are applicable to other public companies that are not emerging growth companies, including not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in this Annual Report on Form 20-F and other periodic reports and proxy statements, extended transition periods for adopting new or revised accounting standards, and exemptions from the requirements of holding a non-binding advisory vote on executive compensation and shareholder approval of any golden parachute payments not previously approved. We will be an emerging growth company until the earliest of: (i) the last day of the fiscal year during which we had total annual gross revenues of $1.07 billion or more, (ii) the last day of the fiscal year following the fifth anniversary of the date of the first sale of the ADSs pursuant to an effective registration statement, (iii) the date on which we have, during the previous three-year period, issued more than $1 billion in non-convertible debt or (iv) the date on which we are deemed a “large accelerated filer” as defined in Regulation S-K under the Securities Act, which means the market value of our ordinary shares that is held by non-affiliates exceeds $700 million as of the prior June 30th. Furthermore, as a foreign private issuer, we are not subject to the same requirements that are imposed upon U.S. domestic issuers by the SEC. Under the Securities Exchange Act of 1934, as amended, or the Exchange Act, we will be subject to reporting obligations that, in certain respects, are less detailed and less frequent than those of U.S. domestic reporting companies. For example, we will not be required to issue proxy statements that comply with the requirements applicable to U.S. domestic reporting companies. We will also have four months after the end of each fiscal year to file our Annual Reports with the SEC and will not be required to file current reports as frequently or promptly as U.S. domestic reporting companies. Furthermore, our officers, directors, and principal shareholders will be exempt from the requirements to report transactions in our equity securities and from the short-swing profit liability provisions contained in Section 16 of the Exchange Act. These exemptions and leniencies, along with other corporate governance exemptions resulting from our ability to rely on home country rules, will reduce the frequency and scope of information and protections to which you may otherwise have been eligible in relation to U.S. domestic reporting companies. See “Item 16G. Corporate Governance Practices” for more information.
37
We cannot predict if investors will find the ADSs less attractive because we may rely on these reduced requirements. If some investors find the ADSs less attractive as a result, there may be a less active trading market for the ADSs and our share price may be more volatile.
Sales of a substantial number of our ordinary shares or ADSs in the public market could cause our share price to fall.
If our existing shareholders sell, indicate an intention to sell, or the market perceives that they intend to sell, substantial amounts of our securities on the Nasdaq Capital Market after the date of this Annual Report on Form 20-F, the market price of our securities could decline significantly. As of March 15, 2019, we had 190,735,668 ordinary shares outstanding. Of those shares, 190,535,668 were freely tradable, without restriction. In addition, we have registered for resale up to 107,191,631 ordinary shares represented by 2,143,833 ADSs, which, subject to there being a current prospectus being available for the resale of those shares, may be sold from time to time in the public markets.
In addition, as of March 15, 2019, an aggregate of 179,328,608 ordinary shares, that are issuable pursuant to exercise of either outstanding options or outstanding warrants or outstanding pre-paid warrants, will become eligible for sale in the public market to the extent permitted by the provisions of various vesting schedules, and Rule 144 and Rule 701 under the Securities Act of 1933, as amended, or the Securities Act. If these additional ordinary shares are sold, or if it is perceived that they will be sold, in the public market, the market price of our ordinary shares could decline.
Future sales and issuances of our securities or rights to purchase securities, including pursuant to our equity incentive plans, could result in additional dilution of the percentage ownership of our shareholders and could cause the prices of our securities to fall.
Additional capital will be needed in the future to continue our planned operations. To the extent we raise additional capital by issuing equity securities, our shareholders may experience substantial dilution. We may sell ordinary shares, ADSs, convertible securities, or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell ordinary shares, ADSs, convertible securities, or other equity securities in one or more transactions, existing investors may be materially diluted by subsequent sales, and new investors could gain rights superior to our existing shareholders.
Pursuant to our Share Ownership and Option Plan (2010), or the 2010 Plan, our management is authorized to grant share options and other equity-based awards to our employees, directors, and consultants. As of March 15, 2019, our officers, directors, employees and consultants hold 55,919,792 options to purchase 35,613,931 ordinary shares under the 2010 Plan. In addition, on January 30, 2019, our board of directors approved the grant of 17,019,500 options to purchase 17,019,500 ordinary shares, which are subject to shareholders approval.
If our board of directors elects to increase the number of shares available for future grant by the maximum amount each year, our shareholders may experience additional dilution, which could cause our share price to fall.
We do not intend to pay dividends on our securities in the foreseeable future, so any returns will be limited to the value of our shares.
We have never declared or paid any cash dividends on our share capital. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. Any return to shareholders will therefore be limited to the appreciation of their shares. In addition, Israeli law limits our ability to declare and pay dividends, and may subject our dividends to Israeli withholding taxes; see “Item 10.B. Memorandum and Articles of Association——Dividend and Liquidation Rights” for additional information. As a result, investors in the ADSs or ordinary shares will not be able to benefit from owning these securities unless their market price becomes greater than the price paid by such investors and they are able to sell such securities. We cannot assure you that you will ever be able to resell our securities at a price in excess of the price paid.
38
In the event we make distributions or dividends, you may not receive the same distributions or dividends as those we make to the holders of our ordinary shares, and, in some limited circumstances, you may not receive dividends or other distributions, or receive any value for them, if it is illegal or impractical to make them available to you.
The depositary for the ADSs has agreed to pay to you the cash dividends or other distributions it or the custodian receives on ordinary shares or other deposited securities underlying the ADSs, after deducting its fees and expenses. You will receive these distributions in proportion to the number of ordinary shares your ADSs represent. However, the depositary is not responsible if it decides that it is unlawful or impractical to make a distribution available to any holders of ADSs. For example, it would be unlawful to make a distribution to a holder of ADSs if it consists of securities that require registration under the Securities Act, but that are not properly registered or distributed under an applicable exemption from registration. In addition, conversion into U.S. dollars from foreign currency that was part of a dividend made with respect to deposited ordinary shares may require the approval or license of, or a filing with, any government or agency thereof, which may be unobtainable. In these cases, the depositary may determine not to distribute such property and hold it as “deposited securities” or may seek to effect a substitute dividend or distribution, including net cash proceeds from the sale of the dividends that the depositary deems an equitable and practicable substitute. We have no obligation to register under U.S. securities laws any ordinary shares, rights, or other securities made available through such distributions. We also have no obligation to take any other action to permit the distribution of ADSs, ordinary shares, rights, or anything else to holders of ADSs. In addition, the depositary may withhold from such dividends or distributions its fees and an amount on account of taxes or other governmental charges to the extent the depositary believes it is required to make such withholding. This means that you may not receive the same distributions or dividends as those we make to the holders of our ordinary shares, and, in some limited circumstances, you may not receive any value for such distributions or dividends if it is illegal or impractical for us to make them available to you. These restrictions may cause a material decline in the value of the ADSs.
Holders of ADSs must act through the depositary to exercise their rights.
Holders of the ADSs do not have the same rights of our shareholders and may only exercise the voting rights with respect to the underlying ordinary shares in accordance with the provisions of the deposit agreement for the ADSs. In general, under Israeli law, the minimum notice period required to convene a shareholders’ meeting is no less than 35 or 21 calendar days, depending on the proposals on the agenda for the shareholders’ meeting. When a shareholders’ meeting is convened, holders of the ADSs may not receive sufficient notice of a shareholders’ meeting to permit them to withdraw their ordinary shares to allow them to cast their vote with respect to any specific matter. In addition, the depositary and its agents may not be able to send voting materials to holders of the ADSs or carry out their voting instructions in a timely manner. We will make all reasonable efforts to cause the depositary to extend voting rights to holders of the ADSs in a timely manner, but we cannot assure holders that they will receive the voting materials in time to ensure that they can instruct the depositary to vote their ADSs. Furthermore, the depositary and its agents will not be responsible for any failure to carry out any instructions to vote, for the manner in which any vote is cast or for the effect of any such vote. As a result, holders of the ADSs may not be able to exercise their right to vote, and they may lack recourse if their ADSs are not voted as they requested. In addition, in the capacity as a holder of ADSs, they will not be able to call a shareholders’ meeting.
You may be subject to limitations on transfer of your ADSs.
Your ADSs are transferable on the books of the depositary. However, the depositary may close its transfer books at any time or from time to time when it deems expedient in connection with the performance of its duties. In addition, the depositary may refuse to deliver, transfer, or register transfers of ADSs generally when our books or the books of the depositary are closed, or at any time if we or the depositary deems it advisable to do so because of any requirement of law or of any government or governmental body or for any other reason in accordance with the terms of the deposit agreement.
39
Your percentage ownership in us may be diluted by future issuances of share capital, which could reduce your influence over matters on which shareholders vote.
Our board of directors will have the authority, in most cases without action or vote of our shareholders, to issue all or any part of our authorized but unissued shares, including ordinary shares issuable upon the exercise of outstanding options and warrants. Issuances of additional shares would reduce your influence over matters on which our shareholders vote.
If equity research analysts do not publish research reports about our business or if they issue unfavorable commentary or downgrade the ADSs, the price of the ADSs could decline.
The trading market for the ADSs will rely in part on the research and reports that equity research analysts publish about us and our business. The price of the ADSs could decline if we do not obtain research analyst coverage or if one or more securities analysts downgrade the ADSs, issue other unfavorable commentary, or cease publishing reports about us or our business.
Risks Related to Our Operations in Israel
We are a “foreign private issuer” and intend to follow certain home country corporate governance practices, and our shareholders may not have the same protections afforded to shareholders of companies that are subject to all corporate governance requirements under the listing rules of the Nasdaq Stock Market LLC, or the Nasdaq Listing Rules.
As a foreign private issuer, we are permitted to follow certain home country corporate governance practices instead of those otherwise required under the Nasdaq Stock Market for domestic U.S. issuers. For instance, we follow home country practice in Israel with regard to the quorum requirement for shareholder meetings. As permitted under the Companies Law, our articles of association provide that the quorum for any meeting of shareholders shall be the presence of at least two shareholders present in person, by proxy, or by a voting instrument, who hold at least 20% of the voting power of our shares. In addition, we will follow home country practices in Israel (and consequently avoid the requirements that would otherwise apply to a U.S. company listed on the Nasdaq Capital Market) with regard to the requirement to obtain shareholder approval for certain dilutive events (such as for the establishment or amendment of certain equity-based compensation plans, issuances that will result in a change of control of the company, certain transactions other than a public offering involving issuances of a 20% or more interest in the company, and certain acquisitions of the stock or assets of another company). We may in the future (or may be required to) elect to follow home country practices in Israel with regard to other matters, as well, such as the formation of compensation, nominating, and governance committees, separate executive sessions of independent directors and non-management directors, amending our compensation policy from time to time, and the approval of certain interested-parties transactions. Following our home country governance practices as opposed to the requirements that would otherwise apply to a U.S. company listed on the Nasdaq Capital Market may provide less protection to you than what is accorded to investors under the Nasdaq Listing Rules applicable to domestic U.S. issuers. See “Item 16G. Corporate Governance Practices” for more information.
In addition, as a foreign private issuer, we are exempt from the rules and regulations under the Exchange Act related to the furnishing and content of proxy statements, including the requirement for an emerging growth company to disclose the compensation of the chief executive officer and other two highest compensated executive officers on an individual, rather than aggregate, basis. Under regulations promulgated under the Companies Law, we will be required to disclose in the notice for our annual meetings of shareholders, the annual compensation of our five most highly compensated officers on an individual basis, rather than aggregate. However, this disclosure will not be as extensive as the disclosure required by a U.S. domestic issuer. We will also have four months after the end of each fiscal year to file our annual reports with the SEC and will not be required to file current reports as frequently or promptly as U.S. domestic reporting companies. Furthermore as a foreign private issuer, our officers, directors and principal shareholders will be exempt from the requirements to report short-swing profit recovery contained in Section 16 of the Exchange Act. Also, as a foreign private issuer, we are not subject to the requirements of Regulation FD (Fair Disclosure) promulgated under the Exchange Act. These exemptions and leniencies will reduce the frequency and scope of information and protections available to you in comparison to those applicable to U.S. domestic reporting companies.
40
In order to maintain our current status as a foreign private issuer, more than 50% of our outstanding voting securities must not be directly or indirectly owned by residents of the U.S., and we must not have any of the following: (i) a majority of our executive officers or directors being U.S. citizens or residents, (ii) more than 50% of our assets being located in the U.S., or (iii) our business being principally administered in the U.S. Although we have elected to comply with certain U.S. regulatory provisions, our loss of foreign private issuer status would make such provisions mandatory. The regulatory and compliance costs to us under U.S. securities laws as a U.S. domestic reporting company may be significantly higher. If we are not a foreign private issuer, we will be required to file periodic reports and registration statements on U.S. domestic reporting company forms with the SEC, which are more detailed and extensive than the forms available to a foreign private issuer. We may also be required to modify certain of our policies to comply with accepted governance practices associated with U.S. domestic reporting companies. Such conversion and modifications will involve additional costs. In addition, we may lose our ability to rely upon exemptions from certain corporate governance requirements on U.S. stock exchanges that are available to foreign private issuers.
Potential political, economic, and military instability in the State of Israel, where the majority of our senior management and our research and development facilities are located, may adversely impact our results of operations.
We are incorporated under Israeli law and our offices and operations are located in the State of Israel. In addition, our employees, officers, and all but three of our directors are residents of Israel. Accordingly, political, economic, and military conditions in Israel directly affect our business. Since the State of Israel was established in 1948, a number of armed conflicts have occurred between Israel and its neighboring countries. Any hostilities involving Israel or the interruption or curtailment of trade between Israel and its present trading partners, or a significant downturn in the economic or financial condition of Israel, could adversely impact our operations. Since October 2000, there have been increasing occurrences of terrorist violence. Ongoing and revived hostilities or other Israeli political or economic factors could harm our operations, product development and results of operations.
Although Israel has entered into various agreements with Egypt, Jordan, and the Palestinian Authority, there has been an increase in unrest and terrorist activity, which began in October 2000 and has continued with varying levels of severity. The establishment in 2006 of a government in the Gaza Strip by representatives of the Hamas militant group has created additional unrest and uncertainty in the region. In 2006, a conflict between Israel and the Hezbollah in Lebanon resulted in thousands of rockets being fired from Lebanon up to 50 miles into Israel. Starting in December 2008, for approximately three weeks, Israel engaged in an armed conflict with Hamas in the Gaza Strip, which involved missile strikes against civilian targets in various parts of Israel and negatively affected business conditions in Israel. In November 2012, for approximately one week, Israel experienced a similar armed conflict, resulting in hundreds of rockets being fired from the Gaza Strip and disrupting most day-to-day civilian activity in southern Israel. Most recently, in July 2014, Israel yet again experienced rocket strikes against civilian targets in various parts of Israel, as part of an armed conflict commenced between Israel and Hamas. If continued or resumed, these hostilities may negatively affect business conditions in Israel in general and our business in particular. Our insurance policies do not cover us for the damages incurred in connection with these conflicts or for any resulting disruption in our operations. The Israeli government, as a matter of law, provides coverage for the reinstatement value of direct damages that are caused by terrorist attacks or acts of war; however, the government may cease providing such coverage or the coverage might not be enough to cover potential damages. In the event that hostilities disrupt the ongoing operation of our facilities or the airports and seaports on which we depend to import and export our supplies and products, our operations may be materially adversely affected.
In addition, since the end of 2010, numerous acts of protest and civil unrest have taken place in several countries in the Middle East and North Africa, many of which involved significant violence. The civil unrest in Egypt, which borders Israel, resulted in the resignation of its president Hosni Mubarak, and to significant changes to the country’s government. In Syria, also bordering Israel, a civil war is continuing to take place. The ultimate effect of these developments on the political and security situation in the Middle East and on Israel’s position within the region is not clear at this time. Such instability may lead to deterioration in the political and trade relationships that exist between the State of Israel and certain other countries.
41
Popular uprisings in various countries in the Middle East and North Africa are affecting the political stability of those countries. Such instability may lead to deterioration in the political and trade relationships that exist between the State of Israel and these countries. Several countries, principally in the Middle East, still restrict doing business with Israel and Israeli companies, and additional countries may impose restrictions on doing business with Israel and Israeli companies if hostilities in Israel or political instability in the region continues or increases. Any hostilities involving Israel, interruption or curtailment of trade between Israel and its present trading partners, or significant downturns in the economic or financial condition of Israel could adversely affect our operations and product development and adversely affect our share price. Similarly, Israeli companies are limited in conducting business with entities from several countries. For instance, in 2008, the Israeli legislature passed a law forbidding any investments in entities that transact business with Iran.
In addition, Iran has threatened to attack Israel and is widely believed to be developing nuclear weapons. Iran is also believed to have a strong influence among extremist groups in the region, such as Hamas in Gaza, Hezbollah in Lebanon, and various rebel militia groups in Syria. Additionally, a violent jihadist group named Islamic State of Iraq and Levant, or ISIL, is involved in hostilities in Iraq and Syria. Although ISIL’s activities have not directly affected the political and economic conditions in Israel, ISIL’s stated purpose is to take control of the Middle East, including Israel. These situations may potentially escalate in the future to more violent events, which may affect Israel and us. Any armed conflicts, terrorist activities, or political instability in the region could adversely affect business conditions and could harm our results of operations and could make it more difficult for us to raise capital. Parties with whom we do business may decline to travel to Israel during periods of heightened unrest or tension, forcing us to make alternative arrangements when necessary in order to meet our business partners face to face. In addition, the political and security situation in Israel may result in parties with whom we have agreements involving performance in Israel claiming that they are not obligated to perform their commitments under those agreements pursuant to force majeure provisions in such agreements. Further, in the past, the State of Israel and Israeli companies have been subjected to economic boycotts. Several countries still restrict business with the State of Israel and with Israeli companies. These restrictive laws and policies may have an adverse impact on our operating results, financial condition, or the expansion of our business.
Our operations may be disrupted by the obligations of personnel to perform military service.
As of March 15, 2019, we had 42 employees, all of whom were based in Israel. Some of our employees may be called upon to perform up to 36 days (and in some cases more) of annual military reserve duty until they reach the age of 40 (and in some cases, up to 45 or older) and, in emergency circumstances, could be called to immediate and unlimited active duty. In the event of severe unrest or other conflict, individuals could be required to serve in the military for extended periods of time. Since September 2000, in response to increased tension and hostilities, there have been occasional call-ups of military reservists, including in connection with the 2006 conflict in Lebanon, and the December 2008, November 2012 and July 2014 conflicts with Hamas, and it is possible that there will be additional call-ups in the future. Our operations could be disrupted by the absence of a significant number of our employees related to military service or the absence for extended periods of one or more of our key employees for military service. Such disruption could materially adversely affect our business and results of operations. Additionally, the absence of a significant number of the employees of our Israeli suppliers and contractors related to military service or the absence for extended periods of one or more of their key employees for military service may disrupt their operations.
The tax benefits that are available to us if and when we generate taxable income require us to meet various conditions and may be prevented or reduced in the future, which could increase our costs and taxes.
If and when we generate taxable income, we may be eligible for certain tax benefits provided to “Preferred Enterprises” under the Israeli Law for the Encouragement of Capital Investments, 5719-1959, as amended, or the Investment Law. The benefits that may be available to us under the Investment Law are subject to the fulfillment of conditions stipulated in the Investment Law. Further, in the future these tax benefits may be reduced or discontinued. If these tax benefits are reduced, cancelled, or discontinued, our Israeli taxable income would be subject to regular Israeli corporate tax rates. The standard corporate tax rate for Israeli companies is currently 23%. Additionally, if we increase our activities outside of Israel through acquisitions, for example, our expanded activities might not be eligible for inclusion in future Israeli tax benefit programs. See “Item 10.E. Taxation—Israeli Tax Considerations and Government Programs—Law for the Encouragement of Capital Investments, 5719-1959.”
42
It may be difficult to enforce a U.S. judgment against us, our officers and directors, and the Israeli experts named in this Annual Report on Form 20-F in Israel or the United States, or to assert U.S. securities laws claims in Israel or serve process on our officers and directors and these experts.
We were incorporated in Israel, and our corporate headquarters and substantially all of our operations are located in Israel. All of our senior management and a majority of our directors are located outside the United States. All of our assets are located outside the United States. Therefore, it may be difficult for an investor, or any other person or entity, to enforce a U.S. court judgment based upon the civil liability provisions of the U.S. federal securities laws against us or any of these persons in a U.S. or Israeli court, or to effect service of process upon these persons in the United States. Additionally, it may be difficult for an investor, or any other person or entity, to assert U.S. securities law claims in original actions instituted in Israel. Israeli courts may refuse to hear a claim based on an alleged violation of U.S. securities laws against us or our officers and directors on the grounds that Israel is not the most appropriate forum in which to bring such a claim. Even if an Israeli court agrees to hear a claim, it may determine that Israeli law and not U.S. law is applicable to the claim. If U.S. law is found to be applicable, the content of applicable U.S. law must be proved as a fact by expert witnesses, which can be a time-consuming and costly process. Certain matters of procedure would be governed by Israeli law. There is little binding case law in Israel addressing the matters described above.
Your rights and responsibilities as our shareholder will be governed by Israeli law, which may differ in some respects from the rights and responsibilities of shareholders of U.S. corporations.
Because we are incorporated under Israeli law, the rights and responsibilities of our shareholders are governed by our articles of association and Israeli law. These rights and responsibilities differ in some material respects from the rights and responsibilities of shareholders of U.S. corporations. In particular, a shareholder of an Israeli company has a duty to act in good faith and in a customary manner in exercising its rights and performing its obligations towards the company and other shareholders and to refrain from abusing its power in the company, including, among other things, in voting at the general meeting of shareholders on certain matters, such as an amendment to the company’s articles of association, an increase of the company’s authorized share capital, a merger of the company, and approval of related party transactions that require shareholder approval. A shareholder also has a general duty to refrain from discriminating against other shareholders. In addition, a controlling shareholder or a shareholder who knows that it possesses the power to determine the outcome of a shareholder vote or to appoint or prevent the appointment of an officer of the company has a duty to act in fairness towards the company with regard to such vote or appointment. However, Israeli law does not define the substance of this duty of fairness. There is limited case law available to assist us in understanding the nature of this duty or the implications of these provisions. These provisions may be interpreted to impose additional obligations and liabilities on our shareholders that are not typically imposed on shareholders of U.S. corporations. See “Item 6.C. Board Practices—Approval of Related Party Transactions under Israeli Law—Shareholders’ Duties.”
Provisions of Israeli law and our amended and restated articles of association could make it more difficult for a third party to acquire us or increase the cost of acquiring us, even if doing so would benefit our shareholders.
Israeli law regulates mergers, requires tender offers for acquisitions of shares above specified thresholds, requires special approvals for transactions involving directors, officers, or significant shareholders and regulates other matters that may be relevant to such types of transactions. For example, a tender offer for all of a company’s issued and outstanding shares, or a Full Tender Offer, can only be completed if the acquirer receives approval of the holders of at least 95% of the issued share capital. Completion of the Full Tender Offer also requires approval of a majority of the offerees that do not have a personal interest in the tender offer, unless at least 98% of the company’s outstanding shares are tendered. Furthermore, the shareholders, including those who indicated their acceptance of the Full Tender Offer (unless the acquirer stipulated in its tender offer that a shareholder that accepts the offer may not seek appraisal rights), may, at any time within six months following the completion of the tender offer, petition an Israeli court to alter the consideration for the acquisition. In case the Full Tender Offer has not been accepted by the required threshold, the offeror is limited to acquire shares that will confer on the offeror a holding of not more than 90% of the issued share capital of the company. See “Item 10.B. Memorandum and Articles of Association—Acquisitions under Israeli Law” for additional information.
43
Further, Israeli tax considerations may make potential transactions undesirable to us or to some of our shareholders whose country of residence does not have a tax treaty with Israel granting tax relief to such shareholders from Israeli tax. For example, Israeli tax law does not recognize tax-free share exchanges to the same extent as U.S. tax law. With respect to mergers, Israeli tax law allows for tax deferral in certain circumstances but makes the deferral contingent on the fulfillment of a number of conditions, including, in some cases, a holding period of two years from the date of the transaction during which sales and dispositions of shares of the participating companies are subject to certain restrictions. Moreover, with respect to certain share swap transactions, the tax deferral is limited in time, and when such time expires, the tax becomes payable even if no disposition of the shares has occurred.
We have received grants from the IIA for certain research and development expenditures. The terms of these grants may require us to satisfy specified conditions in order to manufacture products and transfer technologies outside of Israel. For more information, see “—Risks Related to Our Financial Condition and Capital Requirements—The IIA grants we have received for research and development expenditures restrict our ability to manufacture products and transfer know-how outside of Israel and require us to satisfy specified conditions.”
We may be classified as a passive foreign investment company for U.S. federal income tax purposes, and our U.S. shareholders may suffer adverse tax consequences as a result.
Generally, if, for any taxable year, either, at least 75% of our gross income is passive income (including our pro-rata share of the gross income of our 25% or more-owned corporate subsidiaries), or at least 50% of the average value of our assets (including our pro-rata share of the assets of our 25% or more-owned corporate subsidiaries) is attributable to assets that produce passive income or are held for the production of passive income, we would be characterized as a passive foreign investment company, or PFIC, for U.S. federal income tax purposes. Passive income generally includes dividends, interest, and gains from disposition of passive assets and rents and royalties.
If we are characterized as a PFIC for any taxable year (or portion thereof) that is included in the holding period of a U.S. holder (as defined below) of our securities, such U.S. holder generally will be subject to certain adverse U.S. federal income tax consequences, including increased tax liability on gains from dispositions of our securities and certain distributions and a requirement to file annual reports with the Internal Revenue Service, or IRS. See “Item 10.E. Taxation—Material U.S. Federal Income Tax Consequences—Passive Foreign Investment Company Consequences.”
Since PFIC status depends on the composition of our income and the composition and value of our assets (which may be determined in large part by reference to the market value of our ordinary shares, which may be volatile) from time to time, there can be no assurance that we will not be considered a PFIC for any taxable year. However, based on our non-passive revenue-producing operations for the year ended December 31, 2018, we do not expect to be a PFIC for our 2018 taxable year. Because the PFIC determination is highly fact intensive, there can be no assurance that we will not be a PFIC in 2019 or any other year.
U.S. investors are urged to consult their own tax advisors regarding the possible application of the PFIC rules. For more information, see “Item 10.E. Taxation—Material U.S. Federal Income Tax Consequences—Passive Foreign Investment Company Consequences.”
Our facilities in Israel are subject to local Business Licensing and Planning and Zoning regulations and we may be subject to fines if not complied with.
Under the Israeli Licensing of Businesses Law, to which our production site and offices and laboratories are subject, operating a business without a license or temporary permit is a criminal offense. We have a business license for our laboratories and offices, in effect until December 31, 2019. We also have a business license for our plant growth and production site at Yessod Hama’ala, in effect until November 3, 2019. In addition, our production sites and laboratories are subject to the Israeli Planning and Zoning Law, which sets provisions and obligations, inter alia, regarding the licensing process for a new building, including building permits, non-conforming use and easements, the supervision over its construction, and the required occupancy permits. According to the Planning and Zoning Law, work or use of land without a permit, where such permit is required, a deviation from the permit granted, or use of agricultural land in violation of the law constitute criminal offenses.
44
| ITEM 4. | INFORMATION ON THE COMPANY |
| A. | History and Development of the Company |
CollPlant is a regenerative medicine company focused on 3D bioprinting of tissues and organs, dermal filers for aesthetics, and on developing and commercializing tissue repair products for orthobiologics, and advanced wound care markets. Our products are based on our rhCollagen (recombinant human collagen) that is produced with CollPlant’s proprietary plant based genetic engineering technology.
Our legal and commercial name is CollPlant Holdings Ltd. We hold all of the issued and outstanding shares of CollPlant Ltd. and have no holdings in other companies. CollPlant Ltd. was incorporated in Israel on August 12, 2004 as a private company limited by shares and began its operations as a technology incubator company under the IIA’s technology incubators program. CollPlant Ltd. owns all of our intellectual property.
We were incorporated in Israel on November 9, 1981 as a private company limited by shares. We became a public company in 1993, when all of our ordinary shares were listed on the TASE. On January 31, 2018, our ADSs commenced trading on the Nasdaq Capital Market under the symbol “CLGN”. The ADSs were quoted on the OTCQX from March 2015 to May 25, 2017, and quoted on the OTCQB from May 26, 2017 to January 30, 2018. Our name has changed several times, but has been CollPlant Holdings Ltd. since May 30, 2010, immediately after the consummation of the merger transaction with CollPlant Ltd. We delisted our ordinary shares from the TASE, and the last date of trading of our ordinary shares was on October 29, 2018.
Our principal offices are located at 4 Oppenheimer, Weizmann Science Park, Rehovot 7670104, Israel, and our telephone number is +972-73-232-5600. Our primary internet address is http://www.CollPlant.com. None of the information on our website is incorporated by reference herein. Puglisi & Associates serves as our agent for service of process in the United States for certain limited matters, and its address is 850 Library Avenue, Suite 204, Newark, Delaware 19711.
We use our website (http://www.CollPlant.com) as a channel of distribution of Company information. The information we post on our website may be deemed material. Accordingly, investors should monitor our website, in addition to following our press releases, SEC filings and public conference calls and webcasts. The contents of our website are not, however, a part of this Annual Report.
We are an emerging growth company, as defined in Section 2(a) of the Securities Act, as implemented under the JOBS Act. As such, we are eligible to, and intend to, take advantage of certain exemptions from reporting requirements that generally apply to public companies, including the auditor attestation requirements with respect to internal control over financial reporting under Section 404 of the Sarbanes-Oxley Act, compliance with new standards adopted by the Public Company Accounting Oversight Board which may require mandatory audit firm rotation or auditor discussion and analysis, exemption from say on pay, say on frequency, and say on golden parachute voting requirements, and reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements. We will be an emerging growth company until the earliest of: (i) the last day of the fiscal year during which we had total annual gross revenues of $1.07 billion or more, (ii) the last day of the fiscal year following the fifth anniversary of the date of the first sale of the ADSs pursuant to an effective registration statement, (iii) the date on which we have, during the previous three-year period, issued more than $1 billion in non-convertible debt, or (iv) the date on which we are deemed a “large accelerated filer” as defined in Regulation S-K under the Securities Act, which means the market value of our ordinary shares that is held by non-affiliates exceeds $700 million as of the prior June 30th.
As a foreign private issuer, we are exempt from certain rules and regulations under the Exchange Act that are applicable to other public companies that are not foreign private issuers. For example, although we intend to report our financial results on a quarterly basis, we will not be required to issue quarterly reports, proxy statements that comply with the requirements applicable to U.S. domestic reporting companies, or individual executive compensation information that is as detailed as that required of U.S. domestic reporting companies. We will also have four months after the end of each fiscal year to file our annual report with the SEC and will not be required to file current reports as frequently or promptly as U.S. domestic reporting companies. We may also present financial statements pursuant to International Financial Reporting Standards, or IFRS, instead of pursuant to U.S. generally accepted accounting principles, or U.S. GAAP. Our senior management, directors, and principal shareholders will be exempt from the requirements to report transactions in our equity securities and from the short-swing profit liability provisions contained in Section 16 of the Exchange Act. As a foreign private issuer, we will also not be subject to the requirements of Regulation FD (Fair Disclosure) promulgated under the Exchange Act.
45
Our capital expenditures for December 31, 2018, 2017 and 2016 amounted to NIS 3.0 million (approximately $800,000), NIS 447,000 (approximately $129,000) and NIS 492,000 (approximately $128,000), respectively. Our purchases of fixed assets primarily include laboratory equipment and establishment of our production site in Rehovot. We financed these expenditures primarily from cash on hand.
| B. | Business Overview |
Overview
We are a regenerative medicine company focused on developing and commercializing tissue repair products, initially for three-dimensional, or 3D, bioprinting of tissues and organs, dermal fillers for aesthetics, orthobiologics and advanced wound care markets. Our products are based on our rhCollagen, a form of human collagen produced with our proprietary plant-based genetic engineering technology. We believe our technology is the only commercially viable technology available for the production of genetically engineered, or recombinant, human collagen. We believe that our rhCollagen, though laboratory-derived, is identical to the type I collagen produced by the human body, has significant advantages compared to currently marketed tissue-derived collagens, including improved biological function, high homogeneity, and reduced risk of immune response. We believe the attributes of our rhCollagen make it suitable for numerous tissue repair applications throughout the human body. We believe that the annual market size for our BioInk, VergenixSTR, VergenixFG and our dermal filler exceeded $8 billion in 2016, and is estimated to reach $12 billion in 2024.
Our rhCollagen has superior biological function when compared to any tissue-derived collagens, whether from animal or human tissues, according to data published in peer-reviewed scientific publications. Our rhCollagen can be fabricated in different forms and shapes including gels, pastes, sponges, sheets, membranes, fibers, and thin coats, all of which have been tested in vitro and in animal models and proven superior to tissue-derived products. We have demonstrated that, due to its homogeneity, rhCollagen can produce fibers and membranes with high molecular order, meaning there is high molecular alignment, which enables the formation of tissue repair products with distinctive physical properties. We produce our rhCollagen in genetically engineered tobacco plants, assuring a relatively abundant supply of high quality raw materials.
Our three leading rhCollagen-based products are:
| ● | CollPlant rhCollagen-based BioInk for use in the 3D printing of tissues and organs. Our flagship BioInk product line provides an ideal building block for three dimensional bioprinting of tissues and organs. The BioInk is being developed to enable the printing of three-dimensional scaffolds combined with human cells and/or growth factors as a basis for tissue or organ formation. In addition to collagen, CollPlant’s BioInk formulations can include other proteins and/or polymers as well. Our BioInk is being developed to be compatible with numerous 3D bioprinting technologies and with printed organ characteristics. In October 2018, we entered into the United License Agreement pursuant to which CollPlant and LB will collaborate in the development of engineered lungs or lung substitutes using our rhCollagen and BioInk. |
| ● | VergenixSTR, a soft tissue repair matrix composed of our rhCollagen and PRP extracted from the patient’s blood. VergenixSTR is intended to accelerate healing in the treatment of tendinopathy, such as in the elbow tendon (for treatment of “tennis elbow”), rotator cuff, patellar tendon, Achilles tendon, and hand tendons. VergenixSTR forms a viscous gel matrix to serve as a scaffold in the vicinity of a tendon injury site. Following the scaffold formation, our rhCollagen activates the platelets in PRP to provide sustained release of growth factors, which promote healing and repair of tendon injuries. In August 2016, we completed an open label, single arm, multi-center clinical trial of VergenixSTR in Israel. In October 2016, we received CE marking certification for VergenixSTR, which is required for a product to be marketed in the European Union. In November 2016, we entered into an exclusive distribution agreement with Arthrex GmbH in Munich, Germany, an affiliate of Arthrex, Inc., for VergenixSTR covering Europe, the Middle East, India, and certain African countries and sales began in Europe. In November 2018, we entered into an exclusive distribution agreement with Joinsmart Biomedical Co. for VergenixSTR in Taiwan. The distributor is currently in the process of certification and authorization of VergenixSTR, In January 2019, the U.S. Food and Drug Administration, or FDA, responded to the Company’s Pre-RFD regarding product classification and jurisdictional assessment. The FDA’s OCP determined that VergenixSTR should be classified as a combination product and should be assigned to the FDA’s CBER. A Pre-RFD is FDA’s preliminary, nonbinding assessment of (1) the regulatory identity or classification of a product as a drug, device, biological product, or combination product, and (2) which FDA Center (i.e., CBER, CDER, or CDRH) will have primary jurisdiction for the premarket review and regulation of the product. Therefore, this classification and jurisdictional assessment is subject to change. We currently do not intend to pursue an FDA regulatory pathway to market for VergenixSTR in 2019. |
46
| ● | VergenixFG, a wound-filling flowable gel made from our rhCollagen. In the European Union, VergenixFG is intended to enhance the quality and speed of closure of deep surgical incisions and wounds, including diabetic ulcers, burns, bedsores, and other chronic wounds. The VergenixFG formulation provides a scaffold that fills the wound site and establishes intimate contact with the surrounding tissue. VergenixFG provides complete coverage of the wound site, facilitates wound closure through an engineered synchronization between scaffold degradation and growth of new tissue, and offers a non-allergenic and pathogen-free scaffold for safe and efficacious wound care therapy. We completed an open label, single arm, multi-center clinical trial of VergenixFG in Israel to support CE marking certification. In February 2016, we received CE marking certification for VergenixFG. Since then we have entered into distribution agreements for the distribution of VergenixFG in 20 countries in Europe, and in Africa. |
Collagen and Collagen-Based Products
Collagen is the main component of connective tissue and is the most abundant protein in mammals. In humans, it comprises approximately 30% of the protein found in the body. Due to its unique characteristics and diverse profile in human body functions, collagen is frequently selected from a variety of biocompatible materials for use in tissue repair to support structural integrity, induce cellular infiltration and promote healing. We estimate that by 2024, the size of the market for human collagen-based tissue repair with our BioInk, VergenixSTR, VergenixFG and our dermal filler for use in 3D bioprinting, aesthetics, orthobiologics and advanced wound care applications is estimated to reach approximately $12 billion.
Type I collagen is the most abundant form of collagen in the human body. It is the dominant constituent of connective tissue and serves as the primary scaffold in tissue or organ repair processes, making it a logical choice for regenerative medicine products. It is found in tendons, skin, artery walls, corneas, the endomysium surrounding muscle fibers, fibrocartilage, and the organic part of bones and teeth. Type II collagen is primarily found in articular cartilage. Type III collagen, which is produced quickly by young fibroblasts before the tougher type I collagen is synthesized, is found in granulation tissue such as artery walls, skin, intestines, and the uterus. While there may be some niche applications in the future where type III or possibly type II collagen is appropriate, type I collagen is best suited for applications associated with regenerative medicine because of its essential role in the healing process of bones, skin, and tendons. Type III recombinant human collagen is currently available for the research market, and is not used in any products currently approved for medical use.
Disadvantages of Current Collagen-Based Products
Currently, type I collagen for medical use is primarily derived from bovine (cow) and porcine (pig) sources, as well as from human cadavers. It is extracted from the tissues using mechanical processes and chemical treatments. Tissue-derived collagens suffer from a number of disadvantages:
| ● | The harsh chemical conditions required to recycle collagen from mature tissue results in a collagen product with random defects in its protein structure, leading to a compromised triple helix. Consequently, tissue-derived collagens have significant damage to binding sites for progenitor cells, which are required for cell proliferation and differentiation into tissue. |
| ● | Tissue-derived collagens are non-homogenous and contains high proportions of cross-linked collagen species with high molecular weight. The rate of degradation of collagen is based on the proportion of cross-linked collagen species within the product. Excessive proportions of cross-linked collagen can impair the collagen’s ability to self-assemble homogenous scaffolds with a high surface area and fully functional integrin-binding capacity, and can also impede its rate of degradation. The inability to effectively control the level of cross-linked collagen species in tissue-derived collagens results in variability of performance for a given product, and affects the rate of infiltration of cells into the scaffold, which can delay healing. |
47
| ● | The extraction of collagen from mature mammalian tissues leaves, in many cases, contaminant proteins, growth factors, and cytokines. As a result, scaffolds made of tissue-derived collagens may provoke inflammation, as well as undesirable immune and foreign body responses that may cause adverse effects and unpredictable biological outcomes. |
| ● | Extraction from animals or humans is also associated with risk of disease transmission. Since 2007, the FDA has highlighted the risks of transmissible diseases to humans in medical devices that contain materials derived from animal sources. In January 2014, the FDA released draft guidance suggesting precautionary procedures to be used in the production of medical devices containing materials derived from animal sources. |
| ● | Although collagen molecules are similar among various animal species, slight differences in the protein sequence between species may result in different biological behavior when applied to humans, and in some cases, invoke specific immune responses; for example, bovine collagen is associated with hypersensitivity and allergic reactions in approximately 3% of people. |
Advantages of our rhCollagen and rhCollagen-based Products
All of our products are based on our proprietary recombinant type I human collagen, rhCollagen, though laboratory-derived, is identical to the type I collagen produced by the human body. The graphic below illustrates the structural differences between rhCollagen produced with our proprietary plant-based technology and currently marketed tissue-derived collagens.
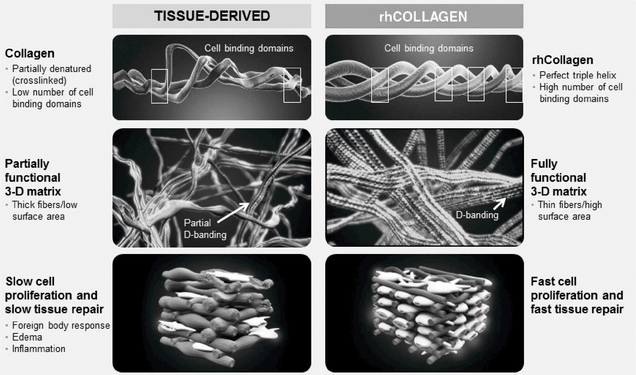
The key advantages of products using our rhCollagen, as compared to those using collagen derived from animals or human cadaveric tissue, include:
| ● | Better biofunctionality in tissue regeneration. Our rhCollagen has superior biological function when compared to animal or human tissue-derived collagen and has a number of useful physical characteristics, including thermal stability, or resistance to decomposition at high temperatures, and a pristine triple helix, according to data published in peer-reviewed scientific publications. The triple helix structure of collagen is formed when two α-1 protein chains and one α-2 protein chain wind together along a common axis. In the formation of rhCollagen, this structure is achieved without modifications that can lead to defects in the triple helix structure in human tissue-derived collagen, hereby leading to a pristine triple helix identical to the form found in nature. A pristine triple helix enables superior binding, which accelerates primary human cell proliferation. Collagen scaffolds of our rhCollagen support endothelial, fibroblast, and keratinocyte cell attachment and proliferation. In all cell types tested, cell proliferation was significantly better in scaffolds made of rhCollagen than in commercially available scaffolds made of bovine collagen. The accelerated cell proliferation achieved with our rhCollagen results in faster wound healing, less scarring, and higher quality tissue regeneration. |
| ● | High homogeneity. Because our rhCollagen is synthesized by five human genes in tobacco plants producing pure molecules that are repeatable and identical to type I human collagen, it is more homogenous than collagen derived from animal or human tissue sources. The high level of homogeneity of our rhCollagen allows the formulation of extremely high concentrations of monomeric, or single-molecule, collagen, up to 150-200mg/ml, which is at least 10 to 100 times higher than the concentration achieved with tissue-derived collagen. The high concentration of homogeneous monomeric collagen is of particular importance where strong collagen fibers are needed for 3-D scaffolds. The homogeneity of our rhCollagen enables us to engineer consistent and reproducible products with a controlled degradation rate which can be optimized to the targeted indication. Achieving the same level of engineered performance would be difficult, if not impossible, with tissue-derived collagen that varies from batch to batch. |
48
| ● | Improved safety and greater purity. Our pure rhCollagen does not induce an immunogenic response, whereas impurities carried over from the source of tissue-derived collagen can lead to immune system rejection. In vitro studies performed under an academic collaboration have demonstrated that rhCollagen incubated with activated THP1-macrophages produces significantly lower levels of inflammatory cytokines when compared with bovine collagen that is similarly incubated. This demonstrates that animal-derived collagen can provoke a foreign body response not seen with rhCollagen, which delays healing and increases scarring. Further, with our rhCollagen, there are no potential side effects in the growth of tissue because there are no residues of growth factors. In addition, with tissue-derived collagen, there is a possibility that the animal or human from which the collagen was produced was infected with a virus, prion, or other pathogen. With our rhCollagen there is no known risk of transmitting diseases and pathogens. |
| ● | Novel applications. Due to our ability to control the protein at the molecular level, it is possible to use our rhCollagen to produce products with unique physical features, as well as high repeatability, which is not possible with tissue-derived collagen. As compared to tissue-derived collagen, rhCollagen membranes have shown better thermal stability, improved tensile strength due to alignment of the collagen fibers, and higher levels of transparency. In addition, rhCollagen can be used to produce high concentration solutions of collagen at low viscosities. The unique properties of our rhCollagen make it an ideal building block for many products that we believe cannot currently be produced using tissue-derived collagen, such as BioInks for 3-D printing, artificial tendons, and transparent ophthalmic products. |
We believe the clinical attributes of our rhCollagen will translate into benefits for patients, payors, and physicians, and will be adopted rapidly by the market once our products receive regulatory approval. We believe the improved biofunctionality of our products could lead to faster recovery, better clinical outcomes, and reduced hospitalization time. Our in vivo studies have shown faster tissue remodeling, faster wound closure, and reduced scarring compared to competing products made from tissue-derived collagen.
The advantages of our rhCollagen outlined above have been demonstrated through in vitro testing and in preclinical animal studies, and are based on the performance of rhCollagen alone. The performance demonstrated in these studies is not necessarily indicative of the performance of our products which contain rhCollagen. We cannot assure you that the same advantages of rhCollagen will be seen in clinical testing of our products containing rhCollagen.
We can produce our rhCollagen cost-effectively and have access to an abundant supply of raw materials. Tobacco is a relatively easy plant to grow, and can be cultivated in a wide range of climates and soils. The tobacco plant is an extremely hardy plant, may be grown in very large volumes and its growth time to reach desired maturity is relatively short (about eight weeks). Under our current production technology, we are able to achieve a cost of goods that allows us to offer products at prices that are competitive with tissue-derived collagen. We are advancing a new production process that will result in labor cost reductions and higher yields, assuring an abundant raw material supply as demand for our rhCollagen increases.
Collagen-based products are already used extensively in the marketplace; therefore, we expect our products will likely be eligible for reimbursement by third-party payors, including government agencies and insurance companies. We believe that the demand for tissue-derived collagen will decrease as the market recognizes the significant advantages of our rhCollagen.
49
Our Market Opportunity
Our rhCollagen represents a platform for the development of products addressing significant opportunities in multiple therapeutic, aesthetic, and other medical markets. We are initially focused on BioInk for use in the 3D printing of tissues and organs, dermal fillers for aesthetics, orthobiologics and advanced wound care markets.
We also see a significant opportunity to use our rhCollagen platform to develop products to address additional indications in these markets as well as in new markets, including cardiovascular, aesthetics to develop next generation of dermal fillers and breast implants, and ophthalmic markets. We believe that the potential addressable market opportunity for products using our technology is even greater than the market size served by currently available collagen-based products, mainly due to continued unmet medical needs and the shortcomings of tissue-derived collagen.
BioInk for 3D printing of tissues & organs
Regenerative medicine and tissue engineering have seen unprecedented growth in the past decade, driving the field of artificial tissue models towards a revolution in future medicine. Progress has been achieved through the development of innovative biomanufacturing strategies to pattern and assemble cells and extracellular matrix, or ECM, in three dimensions to create functional tissue constructs. Bioprinting has emerged as a promising 3D biomanufacturing technology, enabling precise control over spatial and temporal distribution of cells and ECM. Bioprinting technology can be used to engineer artificial tissues and organs by producing scaffolds with controlled spatial heterogeneity of physical properties, cellular composition, and ECM organization. This innovative approach is increasingly utilized in biomedicine, and has potential to create artificial functional constructs for drug screening and toxicology research, as well as tissue and organ transplantation.
Grand View Research Inc. in a report published in March 2018 notes that the global 3D bioprinting market size was valued at $682 million in 2016 and that the global market was expected to reach $2.6 billion by 2024. The growth of the global market is largely driven by increasing large demand of tissues and organs for transplantation and the innovations and advancements in technology for 3D bioprinting. A large number of people across the globe are waiting for an organ or tissue transplant, due to the large gap in demand for organ transplants and donors. This has created traction in the 3D bioprinting industry for developing live tissues and organs. Different companies along with academic institutes and laboratories are investing capital for 3D bioprinting research and development. Some of the other factors driving the growth of the global market include increasing research and development activities and increasing compliance for 3D bioprinting in drug discovery processes. Growing stem cell research and increasing adoption of 3D bioprinting in cosmetic industry are expected to create ample growth opportunities for the global market.
Orthobiologics Market
The global orthopedic device market size is expected to reach USD 47.7 billion by 2026, according to the study performed by Grand View Research, Inc. and published in 2018. The market is anticipated to expand at a CAGR of 3.1% over the forecast period. An aging population, active demographics, innovative technology, and emerging geographic areas are expected to continue to drive growth in the global orthopedic market. Top market segments within orthopedics include reconstructive devices, such as joint replacements; spinal implants and instruments, used to treat joint pain; fracture repair, including the use of plates and screws; and arthroscopy and soft tissue repair, primarily for sports and movement related injuries.
Chronic complex musculoskeletal injuries that are slow to heal pose challenges to physicians and patients alike. Orthobiologics use cell-based therapies and biomaterials to help injuries heal more rapidly with a superior outcome. These products are made from substances that are naturally found in the body, which dynamically interact with the musculoskeletal system to facilitate the healing of bone, cartilage, meniscus, tendons, and ligaments affected by disease or injury. Orthobiologics products are spread across all segments of the larger orthopedic market, generating much of the growth within orthopedics. In 2018, market report issued by Credence Research, Inc. “Orthobiologics Market – Growth, Future Prospects, Competitive Analysis, 2018 – 2026,” the global Orthobiologics market was expected at $4.9 billion in 2017 increasing at a CAGR of 5.7% from 2018 to 2026.
The orthobiologics market is segmented as follows:
| ● | Cell-based therapies, such as PRP; |
| ● | Bone allografts; |
| ● | Bone graft substitutes; |
| ● | Viscosupplementation; and |
| ● | Growth factors, such as BMP. |
50
It is estimated that bone and joint disorders account for approximately half of all chronic conditions in individuals above 50 years of age in developed countries, and they are the most common cause of severe, long-term pain and disability. Moreover, the U.S. population aged 60 years and above is projected to increase by 33% this decade, which represents a key driver of this market as elderly patients are slower to heal and more in need of products that enhance and speed recovery. A rise in the geriatric population along with lifestyle changes such as increased obesity and growing participation in sports and outdoor activities among the older as well as younger generation all contribute to the increase in musculoskeletal disorders.
Advanced Wound Care Market
The global market for wound care encompasses traditional dressings and bandages, as well as advanced wound care products such as bioengineered skin and skin substitutes and wound care growth factors. Over the past 30 years, there has been a shift from traditional wound dressings towards advanced therapies that aim to optimize the wound healing environment. Advanced wound care is composed of biocompatible products that are intended to actively promote wound healing by interacting either directly or indirectly with wound tissues. Attempts to reduce the duration of hospital stays in order to limit healthcare costs and the goal of enhancing therapeutic outcomes are driving the demand for advanced wound care and closure products. One of the primary market drivers for advanced wound care products is the increasing incidence of chronic wounds, which are on the rise due to an aging population and a sharp rise in the incidence of diabetes and obesity worldwide. Both advanced age and chronic medical conditions are associated with a slower healing process, and all phases of wound healing are affected. The inflammatory response is decreased or delayed, as is the proliferative response.
The global advanced wound care market is expected to reach $11.5 billion in 2025 from $8.2 billion in 2017. The market is estimated to grow with a CAGR of 4.5% from 2016-2025, according to ResearchAndMarkets July 2018 report. The three major market segments are device-based wound care, comprised of negative-pressure wound therapy and hydrosurgery systems; moist wound care, comprised of dressings that create and maintain a moist environment; and biologics, comprised of bioactive technologies that provide new approaches to debridement and dermal repair and regeneration.
With a wide range of dressings to choose from, dressing selection is a significant challenge for wound care clinicians. The ideal dressing should induce rapid healing at reasonable cost with minimal inconvenience to the patient. In a healing wound, a cascade of events occurs that includes platelet accumulation, inflammation, fibroblast proliferation, cell contraction, angiogenesis, and re-epithelization, ultimately leading to scar formation and wound remodeling. Collagen plays an important role in each of these phases of wound healing. Native intact collagen provides a natural scaffold or substrate for new tissue growth. Dressings containing collagen are thought to provide the wound with an alternative collagen source that is degraded over time, leaving the endogenous native collagen to continue normal wound healing.
Biological wound dressings have the benefit of forming part of the natural tissue matrix and some of them play an important role in natural wound healing and new tissue formation. These characteristics make them the most attractive and fastest growing segment of the overall advanced wound care market with anticipated double digit growth in upcoming years. In certain instances, these bioactive matrices are incorporated with compounds such as growth factors and antimicrobials for delivery to the wound site. There are a number of biological wound care dressings available that incorporate tissue-derived collagen to enhance wound bed preparation.
51
Our Strategy
We plan to exploit the unique characteristics of our rhCollagen to develop and commercialize an extensive portfolio of regenerative medicine products. The key elements of our strategy include the following:
| ● | Position our rhCollagen as the “gold standard” platform technology for collagen-based products in a broad range of markets. We believe that our rhCollagen represents a significant advance in collagen technology, demonstrated by its biofunctionality, high homogeneity, and reduced risk of immune response. Our rhCollagen is a platform technology which can be utilized in a broad range of therapeutic, aesthetic, and other medical applications, as well as in emerging industries such as 3D bioprinting which we believe cannot be adequately addressed with currently available collagen technologies. We intend to expand the awareness of rhCollagen through partnerships and collaborations with leading commercial and academic partners around the world and further clinical trials which we will seek to have published in peer-reviewed journals, as well as through our participation in academic and industry conferences, to position rhCollagen as the “gold standard” platform technology for collagen-based products. We believe our platform technology, and the knowledge and expertise we have gained in its development, will enable the development, both independently and with collaborators, of differentiated products in multiple industries with a short time to market. |
| ● | Establish a regulatory process for rhCollagen-based end products using VergenixSTR and VergenixFG as precedent. We have obtained marketing clearance for our initial products, VergenixSTR and VergenixFG, through CE marking in Europe. The CE mark is a symbol that indicates that a product conforms with all applicable EU requirements and, once affixed, enables a product to be sold within the European Union and other countries that recognize the CE mark, subject to compliance with applicable submission and approval requirements in such countries. We believe this strategy will allow us to gain earlier market access and thereby more rapid industry acceptance for our rhCollagen-based end products, since the timeline to achieve CE marking is generally shorter than the FDA approval route. Utilizing this strategy is expected to result in more physicians gaining exposure to rhCollagen-based products sooner. We are conducting post-marketing surveillance studies of our products, resulting in physicians gaining more hands-on experience with rhCollagen. Should these post-marketing surveillance studies successfully demonstrate the efficacy of our product, we will endeavor to have these results published in peer-reviewed medical journals as a means of expanding the clinical credibility of rhCollagen and rhCollagen-based end products. With respect to VergenixSTR, in January 2019, the FDA responded to the Company’s Pre-RFD regarding product classification and jurisdictional assessment. The FDA’s OCP determined that VergenixSTR should be classified as a combination product and should be assigned to the FDA’s CBER. A Pre-RFD is FDA’s preliminary, nonbinding assessment of (1) the regulatory identity or classification of a product as a drug, device, biological product, or combination product, and (2) which FDA Center (i.e., CBER, CDER, or CDRH) will have primary jurisdiction for the premarket review and regulation of the product. Therefore, this classification and jurisdictional assessment is subject to change. We currently do not intend to pursue an FDA regulatory pathway to market for VergenixSTR in 2019. |
| ● | Utilize collaborative partners and distributors to develop and commercialize our technology and products. We believe the market-leading characteristics of our rhCollagen will create attractive collaboration opportunities for our products, and we intend to selectively establish collaborations and strategic partnerships with respect to our current and future products in order to accelerate their development and commercialization. We intend to create a commercial organization, initially in Europe, with well-established companies whose distribution networks are deeply entrenched. Our commercial organization will be comprised of the distribution networks of our collaboration partners, particularly in the United States and China, as well as local and regional distributors in certain markets. |
| ● | Manufacturing capacity to support commercialization of rhCollagen-based end products. We cultivate the tobacco plants used in the production of our rhCollagen in a network of farms in Israel, and we extract the raw materials used to manufacture our rhCollagen from these tobacco plants. In April 2018, we announced the opening of a manufacturing facility in Israel that is supporting our current commercial needs to manufacture commercial quantities of our rhCollagen and our BioInk in a cost-competitive manner for application in both premium and commodity markets. |
52
| ● | Expand our pipeline through ongoing development of new products. We intend to continue to develop additional products, both independently and with strategic collaborators, initially in 3D bioprinting of tissues and organs, dermal fillers for aesthetics, orthobiologics and advanced wound care markets and subsequently in other high value markets, based on our rhCollagen. Our product pipeline and our research and development program are expected to yield new products in the coming years. Some of these new products are derivatives of current products, and therefore may benefit from an easier regulatory pathway and shorter time to market, should our current products receive FDA regulatory approval. |
| ● | Advance our leadership position in recombinant protein production through our plant-based technology. We continually seek to expand our knowledge of plant-based protein production systems and introduce improvements into our process. We are shifting production to an enhanced line of tobacco plants with higher collagen yield, along with improvements in the growing and cultivation process as well as collagen extraction and purification. As tissue engineering and regenerative medicine continue to evolve and expand, we expect that the demand for high-quality biomaterials will grow. |
Our Products
BioInk for 3D printing of tissues & organs
3D bioprinting is being applied to the field of regenerative medicine to address the need for complex scaffolds, tissues, and organs that are suitable for transplantation. We have developed rhCollagen-based BioInks that are optimized and provides an ideal building block for the three-dimensional bioprinting of tissues and organs.
For that purpose, rhCollagen was modified chemically to adapt the biological molecules for printing such that BioInks keep a controlled fluidity during printing and cure to form hydrogels when irradiated by certain light sources ranging from UV to visible light. The unique viscosity and shear thinning properties of the modified rhCollagen enable the formulation of BioInks that are suitable for different printing technologies including extrusion, ink-jet, Laser Induced Forward Transfer and Stereolithography. The control of chemical modification in combination with illumination energy allows tight control of the physical properties of the resulting scaffolds to match natural tissue properties, from stiff cartilage to soft adipose. BioInks formulated from rhCollagen were evaluated with all major currently available printing technologies and exhibited the required physical properties and excellent support for cells including a series of primary and differentiated human cells.
We believe our BioInk offers ideal characteristics for 3D bioprinting, including:
| ● | Biocompatibility—supports cell viability and promotes proliferation (e.g. endothelial cells, fibroblasts, keratinocytes, MSCs) |
| ● | Potential safety—has not shown to promote allergic and other tissue reactions |
| ● | Optimized viscosity and gelation kinetics—printability and compatibility with multiple printing technologies |
| ● | Curing with a range of light sources based on specific requirements |
| ● | Controlled degradation profile |
| ● | Controlled rheological properties (e.g. viscosity) |
| ● | Shear thinning properties – compatible with inkjet technology |
| ● | Convenient handling at broad range of temperatures and pH (e.g., maintains liquid properties at RT and above –no gelation) |
| ● | Compatible with different photoinitiators to cover the spectrum of 280-500nm |
| ● | Customized physical properties of the printed constructs that are compatible with natural tissues |
53
We have initiated several research collaborations with biotechnology and medical device companies, as well as academic and research institutions. These collaborations include development of technology for 3D bioprinting of life-saving organs and different tissues, such as cornea, using our BioInk formulations. Our collaborations are generally structured such that our partners provide research funding and purchasing of our BioInk to cover the scope of work, in part or in full. This funding is typically reflected as collaboration revenues in our financial statements. Upon entering into a collaboration, we disclose the financial details only to the extent that they are material to our business and not subject to confidentiality agreements with our partners. Research collaborations with academic or research institutions typically involve both us and the academic partner contributing resources directly to projects, but also may involve sponsored research agreements where we fund specific research programs.
In May 2017, we created a division focused on development of our rhCollagen-based BioInk, following the expansion of our research activities in the field of 3D biologic printing of organs and tissues.
In October 2017, we entered into a work plan with one of the world’s leading medical device companies to develop a prototype of 3D-printed orthopedic implant based on our rhCollagen-based BioInk, and collaboration on this is continuing.
On October 19, 2018, CollPlant entered into the United License Agreement with LB, pursuant to which CollPlant and LB will collaborate in the development of engineered lungs or lung substitutes using our rhCollagen and BioInk. Pursuant to the United License Agreement, CollPlant granted to LB and its affiliates, an exclusive, perpetual, royalty-bearing and transferable license of our technology relating to the production and use of our rhCollagen and BioInk for the commercialization of engineered lungs or lung substitutes using 3D bioprinting processes throughout the universe. Further, under the United License Agreement, CollPlant granted to LB and its affiliates, a two-year option to extend the license to engineered organs or organ substitutes of up to three additional organs specified in the United License Agreement (each an “Option Product” and together with lungs, the “Covered Products”). Further, at the end of the option period, LB and its affiliates shall have a one-year right of first refusal to receive an exclusive license under our technology relating to the production and use of our rhCollagen and BioInk for the Option Products. Other than under the United License Agreement, CollPlant has agreed not to conduct, enable or fund any research, development or commercialization, or grant any license, with respect to the Covered Products during the term of the United License Agreement, unless with respect to any Option Product, the option is not exercised and the right of first refusal period expires.
The United License Agreement provides that LB will purchase our BioInk on a non-exclusive basis for use in the development and manufacture of Covered Products and for up to the first two years of the United License Agreement, CollPlant will supply LB with a specified limited quantity of BioInk without charge. The United License Agreement further provides that following effectiveness, LB will build a facility, or engage a manufacturer to build a facility, in the U.S. for the manufacture of our rhCollagen and BioInk and the parties have agreed to collaborate in the design and construction of the facility.
The United License Agreement provides for the payment of an upfront cash payment of $5 million to CollPlant, which was paid to us in November 2018 following effectiveness of the United License Agreement. In addition, the United License Agreement provides for a one-time non-refundable option payment of $3 million per Option Product ($9 million in the aggregate), and up to $30 million of milestone payments payable as follows: (i) $5 million upon completion of the U.S. facility design, (ii) $5 million upon completion of production of a specified amount of BioInk, and (iii) $5 million for FDA marketing approval for each Covered Product (up to $20 million in the aggregate). Further, CollPlant shall be entitled to a fixed-fee royalty payment (subject to certain adjustment) for each product commercially sold during the term of the Agreement, a fee for the supply of certain quantities of BioInk to LB, and reimbursement for certain costs related to the U.S. facility and any payment made by CollPlant to the IIA.
Unless earlier terminated, the United License Agreement will continue in effect on a Covered Product-by-Covered Product and country-by-country basis until the later of (i) the expiration, invalidation or abandonment of the last CollPlant patent covering a Covered Product in a particular country, and (ii) 12 years from the first commercial sale of such Covered Product in such country. Following expiration (unless earlier terminated), the licenses granted to LB in the Agreement will continue on a fully paid-up, irrevocable basis. The United License Agreement may be terminated early by either party for material breach or bankruptcy. In addition, CollPlant may terminate the United License Agreement in the case of a challenge made by LB, its affiliates or sub-licensees with respect to a CollPlant patent covering a Covered Product or if LB and its affiliates and sub-licensees discontinue development and commercialization of all Covered Products for at least one year. LB may terminate the United License Agreement at any time upon 30 days’ written notice with respect to the entirety of the United License Agreement and upon 30 days’ written notice with respect to its license and other rights under the United License Agreement relating to one or more CollPlant patents, on a patent-by-patent and country-by-country basis.
54
VergenixSTR—Tendinopathy Treatment
VergenixSTR is a soft tissue repair matrix that combines cross-linked rhCollagen with PRP, a concentrated blood plasma that contains high levels of platelets, a critical component of the healing process. Platelets contain growth factors that are responsible for stimulating tissue generation and repair, including soft tissue repair, bone regeneration, development of new blood vessels, and stimulation of the wound healing process. VergenixSTR serves as a scaffold to support cell proliferation and the release of growth factors. The product is injected into the affected area and forms a viscous gel matrix which serves as a temporary reservoir for PRP in the vicinity of a tendon injury site, holding the platelet concentrate in place at the injured area. The matrix formed has the capabilities to activate the platelets in PRP, thereby releasing growth factors in a controlled manner and controlled biodegradation time, enabling optimal healing.
The following graphic illustrates the VergenixSTR kit and application:
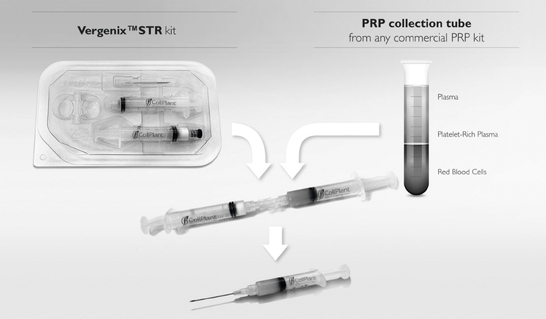
Market for Tendinopathy Treatment
In the European Union, VergenixSTR is intended for the treatment of tendinopathy by promoting healing and repair of tendon injuries in a variety of tendons including the elbow tendon (for treatment of “tennis elbow”), rotator cuffs, patellar tendons, Achilles tendon, and hand tendon.
Tendinopathy: Annual procedures per indication in the United States
Today, the main treatments offered for tendinopathy are local steroid injection, shock wave therapy, and PRP alone. PRP is an orthobiologic that has recently gained popularity as an adjuvant treatment for musculoskeletal injuries. PRP has found application in diverse surgical fields to enhance bone and soft-tissue healing by placing high concentrations of autologous platelets at the site of tissue damage. The platelets contain alpha granules that are rich in several growth factors and play key roles in tissue repair mechanisms. The relative ease of preparation, applicability in the clinical setting, favorable safety profile, and possible beneficial outcome make PRP a promising therapeutic approach for regenerative treatments. One of the challenges in utilizing PRP for tissue repair is the localization of the platelets in the vicinity of the injured tissue. PRP injected alone displays a tendency to migrate and is rapidly degraded. Without addressing the issue of platelet localization, PRP’s efficacy will be limited, particularly in joints like the knee and shoulder which contain relatively large volumes of synovial fluid. VergenixSTR was developed to overcome these inherent limitations associated with the current use of PRP.
We estimate the size of the target market for VergenixSTR for treating tendinopathy is three million procedures per year, or approximately $2.0 billion. While our initial focus for VergenixSTR is in tendinopathy, VergenixSTR may be applicable to other soft tissue indications such as tendon rupture, meniscus tear, and cartilage repair, as well as in the aesthetic market. Transparency Market Research valued the global orthopedic soft tissue market at $5.6 billion in 2013. Globally, the aging population is playing a major role in increasing the incidence of sports injuries as the reduced flexibility and mobility associated with aging can make the body more prone to injury. Consequently, Transparency Market Research forecasts that the orthopedic soft tissue market will grow to $8.5 billion in 2019, a CAGR of 7.2%. The difficulties associated with healing in an aging population highlight the need for advanced orthobiologics products to serve this market.
55
VergenixSTR Product Development
As part of the VergenixSTR development, we conducted a number of preclinical studies to validate the treatment protocol and confirm the enhanced healing potential of the treatment. We completed a preclinical study in August 2013 based on an established model of tendinopathy induced in rats by injection of collagenase into the Achilles tendon. The purpose of this study was to demonstrate the healing ability of VergenixSTR in the treatment of injured and inflamed tendons. The control group participating in the VergenixSTR testing was treated with an injection of PRP only. The efficacy of the product was assessed by histology, measuring parameters of healing at different stages. The preclinical study findings demonstrated that VergenixSTR resulted in lower initial inflammatory mononuclear cell levels, which is known to correlate with a reduction in pain. This effect, along with observations on the appearance of mature fibrosis and elimination of early granulated tissue, suggests that VergenixSTR may accelerate the healing of tendons in comparison with the control treatment.
In a follow-up preclinical study, the ability of VergenixSTR to form a scaffold which is retained to promote healing was assessed through injection of the product into a subcutaneous pocket in rats. Animals treated with VergenixSTR demonstrated a slow degradation of the clot over a period of four to eight weeks, whereas the control group demonstrated nearly immediate dispersion of the injected material.
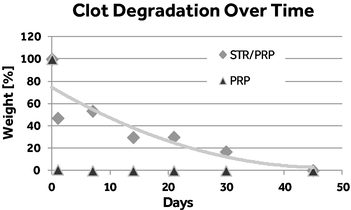
Results of subcutaneous clot implantation in rats. Clot degradation profile is presented as % of weight at time 0.
Analysis of the injection sites showed significant levels of the growth factors PGDF and VEGF, which corresponds to the healing process, throughout the study period suggesting that VergenixSTR is effective in retaining platelet-related growth factors at the site of tendon injury. The preclinical study results appear to confirm VergenixSTR’s ability to promote an improved healing process through the activity of platelet-related growth factors.
We completed a 40 patient open label, single arm, multi-center clinical trial of VergenixSTR at hospitals in Israel which demonstrated the safety and evaluated the performance of VergenixSTR in patients suffering from tennis elbow or lateral epicondylitis. Tennis elbow is an inflammation of the tendons that join the forearm muscles on the outside of the elbow. The forearm muscles and tendons become damaged from overuse, leading to pain and tenderness on the outside of the elbow. Tennis, racquet sports and other sports and activities are a common cause of this condition. Tennis elbow affects 1% to 3% of population in the United States and Europe.
The trial, which commenced in January 2015, initially enrolled 20 patients and was expanded to enroll an additional 20 patients. Patients enrolled in the trial received a one-time injection of VergenixSTR and are monitored for the level of pain, tendon healing, and recovery of hand movement at three and six months after treatment.
In August 2016, we announced final results. At the three-month and six-month follow ups, patients treated with VergenixSTR reported an average 51% and 59% reduction in pain and improvement in motion, respectively, as measured by score improvement over the baseline on the Patient-Rated Tennis Elbow Evaluation, or PRTEE, questionnaire. The PRTEE questionnaire is designed to measure reduction in pain and recovery of motion for patients with tennis elbow. Furthermore, at three-month and six-month follow ups, 74% and 86%, respectively, of patients treated with VergenixSTR showed at least a 25% reduction in pain and improvement in motion as measured by PRTEE. In contrast, a study of standard-of-care tennis elbow therapies published in 2010 in the American Journal of Sports Medicine, or AJSM, reported that, at three and six months, 48% and 36%, respectively, of steroid patients showed at least a 25% reduction in pain and improvement in motion as measured by PRTEE. Also at the three-month and six-month follow ups, 62% and 64%, respectively, of patients treated with VergenixSTR showed at least a 50% reduction in pain and improvement in motion as measured by PRTEE, whereas the 2010 AJSM study showed 33% and 17% reductions at three and six months, respectively, for this same measurement.
56
In October 2016, we received CE marking certification for VergenixSTR. In January 2019, the FDA responded to the Company’s Pre-RFD regarding product classification and jurisdictional assessment. The FDA’s OCP determined that VergenixSTR should be classified as a combination product and should be assigned to the FDA’s CBER. A Pre-RFD is FDA’s preliminary, nonbinding assessment of (1) the regulatory identity or classification of a product as a drug, device, biological product, or combination product, and (2) which FDA Center (i.e., CBER, CDER, or CDRH) will have primary jurisdiction for the premarket review and regulation of the product. Therefore, this classification and jurisdictional assessment is subject to change. We currently do not intend to pursue an FDA regulatory pathway to market for VergenixSTR in 2019. In November 2016, we entered into an exclusive distribution agreement with Arthrex GmbH in Munich, Germany, an affiliate of Arthrex, Inc, for VergenixSTR covering Europe, the Middle East, India, and certain African countries. Sales in Europe commenced in the fourth quarter of 2016. In November 2018, we entered into an exclusive distribution agreement with Joinsmart Biomedical Co. for VergenixSTR in Taiwan. The distributor is currently in the process of certification and authorization of VergenixSTR, as required in Taiwan.
In June 2017, we announced the first positive feedback from treatments as part of our product launch of VergenixSTR in Europe through Arthrex for the treatment of tendinopathy. VergenixSTR was used in treating 45 patients suffering from various cases of tendinopathy including tennis elbow, Achilles tendon, shoulder tendon and plantar fasciitis. Feedback from patient surveys indicated a recovery characterized by a decrease in the level of pain and an improvement in range of motion.
In March 2018, Arthrex announced results of ACP Tendo, a product for treatment of tendinopathy combining our Vergenix®STR and Arthrex’s platelet reach plasma extraction kit, in a European case series. The safety and performance of ACP Tendo was evaluated for the treatment of tendinopathy in 24 patients in 9 different European locations. The indications included injuries in rotator cuff, Achilles tendon, perneal tendon, tibialis tendon and common extensor tendon. In all treatment groups, patient-recorded-pain decreased after 2 weeks and continued along this trend up to the last follow-up at 6 months. Specifically for rotator cuff and common extensor tendon groups, the functionality was increased over the study period, almost achieving pre-symptom levels after 6 months.
VergenixFG—Wound Filler
VergenixFG is an advanced wound care product based on our rhCollagen. In the European Union, VergenixFG is intended for the treatment of deep surgical incisions and deep wounds, including diabetic ulcers, venous and pressure ulcers, burns, bedsores, and other chronic wounds that are difficult to heal. VergenixFG is designed to be easy to use and to be administrated through a cannula by a doctor or nurse. The VergenixFG formulation provides a scaffold of pure human collagen, an important characteristic in promoting the closure of wounds, that fills the wound bed and is engineered to create maximal contact with the surrounding tissue, which is believed to enhance healing. VergenixFG provides complete coverage of the wound site, facilitates wound closure through an engineered synchronization between scaffold degradation and growth of new tissue, and offers a non-allergenic and pathogen-free scaffold for safe and efficacious wound care therapy. Other flowable gel products are available on the market, but they are based on tissue-derived collagen.
Market for Chronic Wounds
VergenixFG is designed to meet the needs of the advanced wound care market, initially in the treatment of chronic wounds. Chronic wounds are rarely seen in individuals who are otherwise healthy. Major chronic diseases such as peripheral vascular diseases, cardiovascular diseases, diabetes, and other debilitating diseases have led to an increase in the incidence of chronic wounds. In wound healing, a cascade of events occurs that includes platelet accumulation, inflammation, fibroblast proliferation, cell contraction, angiogenesis, and re-epithelization, ultimately leading to scar formation. A chronic wound is stalled at one of these healing stages. This usually occurs during the inflammatory phase and is linked to elevated levels of the matrix metalloproteinases, or MMPs, in the wound. During normal wound healing, proteases such as MMPs are attracted to the wound during the inflammatory phase and have an important role in breaking down unhealthy ECMs so that new tissue forms. However, when MMPs are present in a wound at elevated levels for a prolonged period of time, this results in the destruction of healthy ECMs, which is associated with delayed wound healing and an increase in wound size. When the excess of MMPs is not balanced by normal physiological processes, alternative methods are required to reduce protease levels in the wound. This suggests a role for dressings containing collagen in the management of wounds where healing is stalled, as dressings containing collagen are thought to provide the wound with an alternative collagen source that can be degraded by the high levels of MMPs as a sacrificial substrate, leaving the body’s native collagen to continue normal wound healing.
57
We plan on selling VergenixFG at a competitive price to the other advanced healing products in the market. Our initial market for VergenixFG in Europe is chronic wounds, which includes diabetic foot ulcers, venous ulcers, and pressure ulcers. Eucomed has reported there are two million chronic wounds annually in the European Union. We also see the opportunity for expansion of VergenixFG beyond chronic wounds into the treatment of deep surgical incisions. The National Center for Health Statistics reported a total of 51.4 million inpatient surgical procedures took place in the United States in 2010, and we believe at least half of those resulted in a major surgical wound that could benefit from an advanced wound closure product such as VergenixFG to facilitate healing. We estimate that the addressable market for the VergenixFG product within the global advanced wound care market is approximately $3.0 billion.
VergenixFG Product Development
As part of our product development of VergenixFG during the years 2011 to 2013, preclinical studies were conducted by an external laboratory under Good Laboratory Practices, or GLPs. The purpose of the studies was to investigate the performance of VergenixFG in the treatment of wounds in large animals in comparison to a competing product produced from bovine collagen. In a cutaneous full-thickness wound pig model, a broadly accepted model for the human healing process, 95% wound closure was observed with VergenixFG at day 21 compared to 68% closure in wounds treated with the benchmark product. Moreover, VergenixFG treatment induced an early angiogenic response and induced a significantly lower inflammatory response than in the control group. The researchers concluded that VergenixFG proved effective in animal wound models and is expected to be capable of reducing the healing time of human wounds.
We have completed an open label, single arm, multi-center registration trial of VergenixFG of 20 patients in Israel to demonstrate safety and to evaluate the performance of VergenixFG in patients with hard-to-heal chronic wounds of the lower limbs. Patients enrolled in the trial, received a single treatment of VergenixFG followed by a four-week follow up. Product performance was examined according to several measures, the main one being the percentage of wound closure achieved. The results were published in February 2019 in Wounds, a peer-reviewed journal focusing on wound care and wound research. The paper, titled, “A Novel Recombinant Human Collagen-based Flowable Matrix for Chronic Lower Limb Wound Management: First Results of a Clinical Trial,” presents data from a previously reported independent study conducted by physicians at several wound care medical clinics and hospitals in Israel. Four weeks following treatment, nine wounds closed completely, fifteen wounds exhibited a greater than 70% closure, and the median wound area reduction was 94%. Only one patient failed to respond to treatment. All patients in the study reported a 50% reduction in pain. Further, no significant device-related adverse events were reported throughout the study.
In February 2016, we received CE marking certification for VergenixFG. Since then we have entered into distribution agreements for the distribution of VergenixFG in 20 countries in Europe and Africa.
In April 2017, we announced positive results from post-marketing surveillance of 10 patients treated with VergenixFG, for the treatment of patients with chronic, hard-to-heal wounds in Europe. An analysis of the results found average wound closure rates of 80% within five weeks of treatment.
In July 2017, we announced that we started treatments of acute and chronic wounds using VergenixFG for the first time in Israel, by a large private wound-treatment center in the Tel Aviv metropolitan area.
Dermal Filler
We are currently developing a new dermal filler product line, addressing the need for more innovative aesthetic products to treat wrinkles, and are advancing collaborations with leading companies in 3D bioprinting in the field of medical aesthetics with the goal of positioning CollPlant as a major player in the medical aesthetics market.
We are looking to base our new product line on the combination of hyaluronic acid, a naturally-occurring, moisture-binding compound, with our plant-based, tissue regenerating rhCollagen. We are also expanding our development scope to the field of surgical incisions, and are currently running the proof of concept study in breast and abdominal surgeries with our VergenixFG product.
In May 2018, we filed a provisional patent application with the U.S. Patent and Trademark Office for photocurable dermal fillers comprised of rhCollagen and hyaluronic acid, for the aesthetics market.
During the first quarter of 2019, we supplied the first material order of our rhCollagen into the aesthetics market.
58
According to Global Market Insights Inc., global dermal filler market will surpass $8.5 billion by 2024. Rising awareness and acceptance regarding several cosmetic procedures in developed and developing regions coupled with increasing disposable income will impel dermal filler market size. Rising awareness about superior benefits coupled with minimal pain involved with the procedure will drive global dermal filler market growth.
The facial line correction application segment accounted for largest market size of $2.5 billion in 2017 and the segment is forecasted to dominate the market throughout the estimation period. An expanding geriatric population across the globe seeking anti-aging and wrinkle treatment is expected to have a high impact on segmental growth. Accelerating demand for numerous beauty enhancement procedures is expected to further support facial line correction segment growth until 2024.
Technology
Our rhCollagen is based upon research conducted by our founder and Chief Scientist, Prof. Oded Shoseyov. We believe our technology is the only viable technology available for the production of recombinant type I human collagen, the most abundant collagen in the human body.
The production of our rhCollagen begins with the creation of genetically engineered cultures that are transferred to selected greenhouses across Israel and continues with the harvesting of tobacco leaves and the processing of such leaves to an extract which then undergoes purification until the completion of the rhCollagen.
Five human genes encoding heterotrimeric type I collagen are introduced into tobacco plants. The three protein chains that make up type I collagen—two α1 protein chains and one α2 protein chain—are encoded by two genes. The other three genes encode the human prolyl-4-hydroxylase (P4Hα and P4Hβ) as well as lysyl hydroxylase 3 (LH3) enzymes. These enzymes are responsible for key post-translational modifications of collagen, and plants co-expressing all five of these vacuole-targeted genes generate intact procollagen. The plants are grown in a greenhouse under strict growing protocols and mature leaves are transported to a protein extraction facility. Upon extraction, pro-collagen is enzymatically converted to atelocollagen using a plant-derived protease. The protein is purified to homogeneity through a cost-effective industrial process taking advantage of collagen’s unique properties that make it soluble at a very low pH.
rhCollagen forms thermally stable triple helix structures which readily fibrillate at natural pH and low sodium chloride concentrations, making it ideal for use in the manufacture of products for tissue repair in the human body. Binding of integrins (transmembrane receptors) presented by the cells to a specific 3D structure on type I collagen fibrils requires a perfect triple helix. This binding is essential for binding and proliferation of cells on tissue repair scaffolds. In a recent study published in the Journal of Biomedical Materials Research Part B: Applied Biomaterials, rhCollagen was compared with acid-solubilized collagen from bovine dermis and pepsin-solubilized collagen from human fibroblast cell culture. Tested samples of the tissue-derived collagens had random fibrillar organization, whereas rhCollagen membranes showed far greater regional fibril alignment and transparency. RhCollagen membranes also showed better thermal stability compared with the tissue-derived collagens. The authors concluded that cross-linked rhCollagen membranes had a superior combination of desirable properties, namely higher transparency, higher thermal and tensile strengths, and adequate hydration.
We have selected tobacco as the medium for production of rhCollagen due to certain attributes of the tobacco plant that provide us with a number of advantages:
| ● | The genetic structure of tobacco is well understood and therefore can be effectively manipulated. |
| ● | We can monitor the effect of weather conditions on the accumulation of proteins in the plants, which allows us to make optimal use of the growing area. We control the growing process in order to maximize yields. |
59
| ● | Because tobacco is not part of the food chain, there are no concerns about cross-contamination of the food supply that could result from genetically modified plants, which eases the regulatory burden. |
| ● | Tobacco plants may be grown in very large volumes and its growth time until reaching the desired maturity is relatively short (about eight weeks). |
We have developed a large portfolio of configurations and composites based on our rhCollagen that are used to create high-quality products, including our three products, as follows:

Our Development Activities
Development History
Our rhCollagen was first developed as a collaboration among several commercial partners and the Hebrew University of Jerusalem, a major academic institution in Israel, under the direction of Professor Oded Shoseyov. Prof. Shoseyov is a faculty member at the Robert Smith Institute of Plant Science and Genetics at the Hebrew University of Jerusalem. The intellectual property was transferred to our wholly owned subsidiary, CollPlant Ltd.
As part of our regulatory strategy, we first developed and achieved a CE marking for a collagen-based non-invasive dressing, VergenixWD. We pursued a CE mark for this product as a predicate product for achieving in 2016 CE marking for our VergenixSTR and VergenixFG product in the European Union.
Between 2013 and 2017, we developed a surgical matrix, a novel resorbable carrier designed to help accelerate bone healing and formation. The surgical matrix is a novel resorbable carrier composed of rhCollagen and synthetic minerals which is intended to be charged with a bone morphogenetic protein developed by Bioventus for use as a bone graft substitute in bone repair indications such as spinal fusion and trauma. The surgical matrix was developed in collaboration with Bioventus. The collaboration ended in March 2017.
In May 2017, we created a division focused on development of collagen-based biological ink, or BioInk, following the expansion of our research activities in the field of 3D biologic printing of organs and tissues. In October 2018, we entered into the United License Agreement pursuant to which CollPlant and LB will collaborate in the development of engineered lungs or lung substitutes using our rhCollagen and BioInk.
In May 2018, we filed a provisional patent application for photocurable dermal fillers comprising rhCollagen and hyaluronic acid, for the aesthetics market. This application represents an integral part of our Company’s strategy to expand the uses for rhCollagen-based BioInk into new, high value markets. The combination of hyaluronic acid, a naturally-occurring, moisture-binding compound, with our plant-based, tissue regenerating rhCollagen is intended to form the basis for a new dermal filler product line aimed at addressing the need for innovative aesthetic products to treat wrinkles.
Future Development
To facilitate efficient development, our management holds annual research and development meetings where they prioritize development projects and determine future products. The prioritization process is based on several factors, including our business plan, commercial potential of the products, time to market, cost of development, feasibility of the project, and our established strategic objectives. We have several development projects which are in different stages of development.
Future Products
We periodically examine the continued development of other collagen-based products that we have conceived. Each one of our current products offers a platform to product derivatives that can address other indications and contribute to our pipeline and revenues. These derivative products include, for example, the potential use of VergenixSTR for cartilage repair and ACL reconstruction applications and the potential use of VergenixFG for the treatment of deep surgical incisions. Through ongoing research we are also pursuing other platforms for our rhCollagen, such as biomaterial coatings in order to reduce foreign body response and tissue adhesion. We are also in discussions with companies in the field, to develop next generation soft tissue fillers.
60
Other Recombinant Proteins
As tissue engineering and regenerative medicine continue to evolve and expand, we expect that the demand for high-quality biomaterials will grow. There are a number of other extracellular proteins such as elastin, fibronectin, and different types of collagen which may be produced through our plant production system. Another protein, Resilin, has been produced using another proprietary technology for the production of recombinant proteins. Resilin is a polymeric rubber-like protein secreted by insects to specialized cuticle regions, in areas where high resilience and low stiffness are required. Combining collagen at the nano-scale with Resilin to produce fibers resulted in super-performing fibers with greater tensile strength and elasticity exceeding that of natural collagen fibers. This composite biomaterial could be used in indications where elasticity, strength, and memory shape properties are required, such as tendons, meniscus, and nucleus polyposis.
Manufacturing, Supply, and Production
The majority of our product research and development work is carried out at our offices and research laboratories in Weizmann Science Park in Ness-Ziona, Israel. The agricultural research and development and extraction activities for our rhCollagen are carried out at our site in the north of Israel.
We work with subcontractors with greenhouses for growing the tobacco plant containing human collagen in several locations in Israel. This tobacco growth occurs year-round and is optimized to the climate conditions in order to achieve the maximum amount of the protein in the leaves. The growers use our protocols and are monitored by our agronomists to ensure their compliance with these protocols. Each grower has the infrastructure that can be scaled-up to accommodate future demand without additional capital expenditures.
We perform the extraction process by which rhCollagen is extracted from the tobacco plants at our manufacturing facility in the north of Israel. The collagen purification process which produces rhCollagen is carried out by dedicated subcontractors spread across Israel. Our rhCollagen-based products are currently manufactured in the United States by a subcontractor using rhCollagen we supply to them under our production protocols.
We currently have the ability to produce sufficient quantities of quality recombinant type I human collagen to support our product development activities and the sales of VergenixFG and VergenixSTR and BioInk in Europe until 2021. Our activities are focused on yield improvement, scale-up, and cost reduction.
While our upstream and downstream processes are quite robust and efficient, we continuously invest in further yield improvement and scalability, in order to reduce costs. In order to increase yield, we plan to increase biomass per growing area by using new genetic derivatives, improvement and optimization of growing techniques, and introduction of online controls. Our next-generation tobacco plants have been created through improved genetics and cross-breeding and produce three times the amount of collagen as our first-generation plants. Shifting our growing process from tissue culture techniques to cultivation of plants from seed, which we implemented, is also streamlining the production process and reduce costs. In addition, increased growing areas will reduce overall cost per harvest. We also plan further process optimization of our extraction process to increase yields.
We have an approved in-house purification capability. The purification facility includes clean rooms, logistics support areas, and dedicated production equipment to support the Company’s production demand for the next few years.
Under the United License Agreement, LB is required to build a facility, or engage a manufacturer to build a facility, in the U.S. for the manufacture of our rhCollagen and BioInk in connection with the United License Agreement. CollPlant and LB have agreed to collaborate in the design and construction of the facility, and the parties are collaborating in order to establish the facility. LB is responsible for the costs associated with the establishment of the facility.
61
Under our current production techniques, we achieve a cost of goods that allow us to offer competitive pricing in the orthobiologics, advanced wound care, and other premium collagen-based products markets. We anticipate that the above-mentioned production enhancements will reduce the production cost of our rhCollagen to a level that will enable us to be competitive in both premium and commodity markets for collagen-based products.
Sales, Marketing, and Distribution
We sell our BioInk and rhCollagen directly to our business partners, collaborators and selective customers. We anticipate that any products we develop in collaboration with a strategic partner or collaborator, such as organs based on our BioInk for 3D bioprinting, will be marketed by the partner’s sales force.
Until the middle of 2016, our only sales of rhCollagen were to different consumers in the research market. We sell our rhCollagen in the research market under the brand name Collage. Sigma-Aldrich Company distributes Collage in the global research market, which includes, among others, academic institutions and hospitals worldwide. The Collage that we sold to Sigma-Aldrich under this framework is intended only for research laboratories (in vitro) and not for preclinical or clinical (in vivo) uses. We ended our agreement with Sigma-Aldrich effective as of December 31, 2018 and since than we sell our rhCollagen to selective customers.
We are marketing and distributing VergenixSTR and VergenixFG in the European market with business partners. In November 2016, we entered into an exclusive distribution agreement with Arthrex GmbH in Munich, Germany, an affiliate of Arthrex, Inc. for VergenixSTR covering Europe, the Middle East, India, and certain African countries. In December 2016, we supplied our first order to Arthrex and since then we are supplying Arthrex shipments upon purchase orders, to support sales in Europe.
In June 2016, we entered into distribution agreement with an Italian company to distribute VergenixFG in Italy, and in July 2016, we supplied our first order. Subsequently since then we signed distribution agreements to distribute VergenixFG in 20 countries in Europe and Africa. We are currently in discussions with additional distributors in Europe for the commencement of sales of VergenixFG in additional European countries. These potential distributors are active in the wound healing markets and have the existing sales infrastructure in place.
We have commenced post marketing surveillance studies for both VergenixSTR and VergenixFG with our European key opinion leaders and physicians in order to generate additional clinical data that demonstrates the efficacy and superiority of our products. The study is intended to facilitate market adoption of our products in Europe, as well as provide additional support for the submission package to other regulatory agencies, such as the FDA.
Our proprietary end products are marketed, and will be marketed, to physicians, hospitals, and clinics. We plan to expand the awareness of rhCollagen and our rhCollagen-based products to the end users through the publication of clinical trial data as well as marketing studies we may conduct, along with participation in academic and industry conferences. We will also market our rhCollagen to companies who are developing products using collagen and that do not compete with our primary end products. We anticipate entering into collaborations or partnerships with these companies where we would supply them with rhCollagen for use in their products in return for royalties.
Competition
We are not aware of any competitors that produce human collagen from plants or that produce recombinant type I human collagen. However, our industry is characterized by rapidly evolving technology and intense competition, and our rhCollagen-based products will compete with several alternative tissue-derived or synthetic products. Adequate protection of intellectual property, successful product development, adequate funding, and retention of skilled, experienced, and professional personnel are among the many factors critical to success in the pharmaceutical industry.
62
Generally, our competitors currently include large fully integrated companies, as well as academic research institutes and companies in various developmental stages that develop alternative sources and forms of collagen and tissue-derived products.
The primary competitors to our BioInk are potential bio-material inks for 3D biological printing, based on tissue-derived collagens. Manufacturers of these products include, among others, Collagen Solutions and Advanced BioMatrix.
Our VergenixSTR product will compete with companies that sell steroid injections and PRP kits, including Biomet Inc., Harvest Technologies Corporation, MTF Sports Medicine, and Arteriocyte Medical Systems Inc.
The primary competitors to our VergenixFG product are products based on tissue-derived collagens. Manufacturers of these products include, among others, Integra Lifesciences Corporation, Wright Medical Technology Inc., Smith & Nephew, Molnlycke, Convatec, Coloplast, and Urgo.
Intellectual Property
Our success depends, in part, on our ability to protect our proprietary technology and intellectual property. We rely on a combination of patent, trade secret, and trademark laws in the United States and other jurisdictions to protect our intellectual property rights. In addition, we rely on proprietary processes and know-how, intellectual property licenses, and other contractual rights, including confidentiality and invention assignment agreements, to protect our intellectual property rights and develop and maintain our competitive position.
Patents
We have a global patent portfolio that is comprised of ten patent families. Almost three dozen of our patent applications have issued as patents or will issue soon, having been allowed by the relevant patent office. We have exclusive ownership of 17 issued patents in our patent family that cover methods of creating collagen-producing plants and two issued patents in our patent family that cover methods of processing recombinant collagen. These issued patents and others that may issue in the future in these patent families, assuming timely payment of annual fees, are expected to expire in 2025. Our patent portfolio also includes patent families that cover production and use of collagen. Our more recently filed patent application, if granted, could provide patent protection for a particular formulation of our rhCollagen and its use in 3D printing until 2038.
In addition, our patent portfolio includes pending applications, some of which are jointly owned with Yissum Research Development Company of the Hebrew University of Jerusalem Ltd., or Yissum, as well as issued patents that are jointly owned with Yissum, which cover production of other biomaterials.
We are not aware of any impediments to the patent applications being granted in the United States or other jurisdictions. However, our patent applications may never issue as patents, and our issued patents and any that may issue in the future may be challenged, invalidated or circumvented.
Trade Secrets and Confidential Information
In addition to patented technology, we rely on our trade secrets and continuing technological innovations to develop and maintain our competitive position. In an effort to protect our trade secrets, we rely on, among other safeguards, confidentiality and invention assignment agreements to protect our proprietary technology, know-how and other intellectual property that may not be patentable or that we believe is best protected by means that do not require public disclosure. For example, we require our employees, consultants and advisors to execute confidentiality agreements in connection with their employment or consulting relationships with us and to disclose and assign to us inventions conceived in connection with their services to us. These agreements also provide that all confidential information developed or made known to the individual during the course of their relationship with us must be kept confidential, except in specified circumstances.
63
Trademarks
We rely on trade names, trademarks and service marks to protect our name brands. Our registered trademarks in several countries include the following: “collage” and “Vergenix.”
Materials Transfer Agreements
We periodically enter into materials transfer agreements with commercial organizations, medical institutions and research and development institutions to transfer materials and products developed by us. These agreements include provisions that are customary for such agreements concerning the permitted use of the transferred material and any results obtained using the material, confidentiality, the rights in the transferred materials and in the results of the research and/or development in which the materials are used, and instructions concerning care and usage of the materials. These agreements may be used as a basis for further cooperation between us and the counterparties.
We may be unable to obtain, maintain, and protect the intellectual property rights necessary to conduct our business and may be subject to claims that we infringe or otherwise violate the intellectual property rights of others, which could materially harm our business. For a more comprehensive summary of the risks related to our intellectual property, see “Item 3.D. Risk Factors.”
Agreement with Yissum Research Development Company of the Hebrew University of Jerusalem Ltd. with respect to our rhCollagen
Under an agreement dated July 13, 2004 among Meytav—Technological Innovation Center Ltd., Yehuda Zafrir Fagin, Yissum, and Prof. Oded Shoseyov (our Chief Scientist), we carried out a research and development project to develop a process for the production of quality human collagen in plants and further developed the resulting products created by us, Professor Shoseyov and Zafrir, for commercial applications. Yissum and Professor Shoseyov have assigned all intellectual property rights developed by Professor Shoseyov and owned by them to us, including the intellectual property rights in connection with the development of the method for production of quality human collagen in plants.
Government Regulation
We are a developer of tissue products which are subject to extensive regulation in the United States, the European Union and other jurisdictions. These regulations govern, among other things, the introduction of new tissue products, the observance of certain standards with respect to the design, manufacture, testing, promotion and sales of the products, the maintenance of certain records, the ability to track devices, the reporting of potential product defects, the import and export of devices, and other matters.
In order to obtain marketing authorization in the United States, we would be subject to extensive regulation by the FDA and other federal, state, and local regulatory agencies. The Federal Food, Drug, and Cosmetic Act, or FD&C Act, the Public Health Service Act, or the PHS Act, and their implementing regulations set forth, among others, requirements for the research, testing, development, manufacture, quality control, safety, effectiveness, approval, labeling, storage, record keeping, reporting, distribution, import, export, advertising, and promotion of our products. A failure to comply with relevant requirements may lead to administrative, civil, or criminal sanctions. These sanctions could include the imposition by the FDA of a clinical hold or other suspension on clinical trials, refusal to approve pending marketing applications or supplements, withdrawal of approval, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties, or criminal prosecution.
Although the discussion below focuses on regulation in the United States, we anticipate seeking approval for the marketing of our products in other countries which have their own regulatory requirements. Generally, our activities in other countries will be subject to regulations that are similar in nature and scope as that imposed in the United States such as medical device approval, quality system requirements, product data and certifications, although there can be important differences and the number and scope of these regulatory requirements are generally increasing.
64
We must obtain approval by comparable regulatory authorities of foreign countries outside of the European Union and the United States before we can commence clinical trials or marketing of our products in those countries. The approval process varies from country to country and the process may be longer or shorter than that required for FDA approval. In addition, the requirements governing the conduct of clinical trials, product licensing, pricing, and reimbursement vary greatly from country to country. In all cases, clinical trials must be conducted in accordance with the FDA’s regulations, commonly referred to as good clinical practices, or GCPs, and the applicable regulatory requirements and ethical principles that have their origin in the Declaration of Helsinki.
Government regulation may delay or prevent testing or marketing of our products and impose costly procedures upon our activities. The testing and approval process, and the subsequent compliance with appropriate statutes and regulations, require substantial time, effort, and financial resources, and we cannot be certain that the FDA or any other regulatory agency will grant approvals for our products or any future product candidates on a timely basis or at all. The policies of the FDA or any other regulatory agency may change and additional governmental regulations may be enacted that could prevent or delay regulatory approval of our products or any future product candidates or approval of new indications or label changes. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative, judicial, or administrative action, either in the United States or abroad.
Approval by Health Authorities
The following is a summary review of the laws and regulations governing our operations. Our products are medical products, and their marketing, once development is complete, is contingent upon approval of the health authorities in every country in which the products will be marketed:
Israel
Our operations are subject to permits from the Ministry of Health on two levels:
| ● | First, the registration of medical devices, importing and marketing the medical devices and accessories, and issuing the documentation necessary for the export of medical devices from Israel are all supervised by the medical accessories and devices unit, or AMR, of the Ministry of Health. |
| ● | Second, pertaining to research and development, clinical trials in humans are subject to the approval of the Helsinki Committee of the institution conducting the trial, which is governed by the Public Health Regulations (Trials in Human Beings), 1980, including all amendments until 1999, or the Trials in Human Subjects Regulations and are conducted in accordance with the Guidelines for Clinical Trials in Human Subjects issued by the Ministry of Health, or the Guidelines, and the guidelines of the Declaration of Helsinki, or any other approval required by the Ministry of Health. According to the Trials in Human Subjects Regulations and the Guidelines, the Helsinki Committee must plan and approve every experimental process that involves human beings. The Helsinki Committee is an institutional committee that acts in the medical institution where the trial is performed and is the body that approves and supervises the entire trial process. In practice, the physician, who is the principal investigator, submits a trial protocol to the committee on behalf of the requesting party. The committee forwards its decisions regarding the requests for clinical trials that were approved by the committee to the manager of the medical institute and the manager has the authority to approve the requests, and in some cases the additional approval of the Ministry of Health will be required. According to the procedure for medical trials in human beings set forth by the Ministry of Health, the Helsinki Committee will not approve performance of a clinical trial, unless it is absolutely convinced that the following conditions, among others, are fulfilled: (i) the anticipated benefits for the participant in the clinical trial and to the requesting party to justify the risk and the inconvenience involved in the clinical trial to its participant; (ii) the available medical and scientific information justifies the performance to the requested clinical trial; (iii) the clinical trial is planned in a scientific manner that enables a solution to the tested question and is described in a clear, detailed, and precise manner in the protocol of the clinical trial, conforming with the Declaration of Helsinki; (iv) the risk to the participant in the clinical trial is as minimal as possible; (v) optimal monitoring and follow-up of the participant in the clinical trial; (vi) the initiator, the principal investigator and the medical institute are capable and undertake to allocate the resources required for adequate execution of the clinical trial, including qualified personnel and required equipment; and (vii) the nature of the commercial agreement with the principal investigator and the medical institute does not impair the adequate performance of the clinical trial. |
65
All phases of clinical trials conducted in Israel must be conducted in accordance with the Trials in Human Subjects Regulations, including amendments and addenda thereto, the Guidelines, and the International Conference for Harmonized Tripartite Guideline for Good Clinical Practice. The Trials in Human Subjects Regulations and the Guidelines stipulate that a medical study on humans will only be approved after the Helsinki Committee at the hospital intending to perform the study has approved the medical study and notified the relevant hospital director in writing. In addition, certain clinical studies require the approval of the Ministry of Health. The relevant hospital director, and the Ministry of Health, if applicable, also must be satisfied that the study is not contrary to the Declaration of Helsinki or to other regulations.
Additionally the Israeli penal code prohibits bribing a foreign public employee in exchange for any action related to such employee’s role, in order to achieve, guarantee, or promote business activities or other business advantage.
In June 2017, we received AMR approval for VergenixFG and started treating patients in Israel. In March 2018, we received AMR approval for VergenixSTR.
United States
The regulatory process of obtaining product approvals and clearances can be onerous and costly. Foreign companies manufacturing medical devices intended for sale in the United States are required to meet the FDA’s regulatory requirements. The FDA does not recognize the regulatory certification provided by governmental authorities of other countries.
Regulation of Combination Products
The FDA has specified a definition for the term “combination product,” which includes: (1) a product comprised of two or more regulated components, e.g., drug/device, biologic/device, drug/biologic, or drug/device/biologic, that are physically, chemically, or otherwise combined or mixed and produced as a single entity; (2) two or more separate products packaged together in a single package or as a unit and comprised of drug and device products, device and biological products, or biological and drug products; (3) a drug, device, or biological product packaged separately that according to its investigational plan or proposed labeling is intended for use only with an approved individually specified drug, device, or biological product where both are required to achieve the intended use, indication, or effect and where, upon approval of the proposed product, the labeling of the approved product would need to be changed, e.g., to reflect a change in intended use, dosage form, strength, route of administration, or significant change in dose; or (4) any investigational drug, device, or biological product packaged separately that according to its proposed labeling is for use only with another individually specified investigational drug, device, or biological product where both are required to achieve the intended use, indication, or effect.
The FDA is divided into various “Centers” by product type such as the Center for Drug Evaluation and Research, or CDER, CBER, or the CDRH. Different Centers review drug, biologic, or device applications.
The FDA is charged with assigning a Center with primary jurisdiction, or a lead Center, for review of a combination product. That determination is based on the “primary mode of action,” or PMOA, of the combination product. Thus, if the PMOA of a device-biologic combination product is attributable to the biologic product, CBER, which is responsible for premarket review of the biologic product, would have primary jurisdiction for the combination product.
The FDA has also established an Office of Combination Products to address issues surrounding combination products and provide more certainty to the regulatory review process. That office serves as a focal point for combination product issues for agency reviewers and industry. It is also responsible for developing guidance and regulations to clarify the regulation of combination products and for assignment of the FDA center that has primary jurisdiction for review of combination products where the jurisdiction is unclear or in dispute.
66
After formally establishing the PMOA through an applicant’s Request for Designation, the Center that regulates that portion of the product that generates the PMOA becomes the lead evaluator. When evaluating an application, a lead Center may consult other centers but still retain complete reviewing authority, or it may collaborate with another Center, wherein the lead Center assigns concurrent review of a specific section of the application to another Center, delegating its review authority for that section.
Typically, the FDA requires a single marketing application submitted to the Center selected to be the lead evaluator, although the agency has the discretion to require separate applications to more than one Center. One reason to submit multiple evaluations is if the applicant wishes to receive some benefit that accrues only from approval under a particular type of application, like new drug product or orphan drug exclusivity. If multiple applications are submitted, each may be evaluated by a different lead Center. When submitting multiple applications, the applicant may be subject to the payment of two user fees, but a waiver of such fees may be obtained under certain limited circumstances.
The FDA may subject a combination product to two or more sets of legal authorities, e.g., drug/device, biologic/device, drug/biologic drug, but it has the authority to deem one set of legal authorities sufficient. FDA’s standard of review for a combination products application and the applicable legal authority or authorities will depend on a case-by-case basis evaluation of the scientific and technical issues and risk profile relevant to a combination product and its constituent parts. Because of the breadth and complexity of this analysis in each case, no single regulatory paradigm is appropriate for all combination products.
After receiving FDA approval or clearance, an approved or cleared product must comply with postmarket safety reporting requirements applicable to the product based on the application type under which it received marketing authorization. In the case of current good manufacturing practices, or cGMP, the applicant may take one of two approaches: (1) complying with cGMP for each constituent part, or (2) a streamlined approach specific to combination products, subject to certain limitations.
In January 2019, the U.S. Food and Drug Administration, or FDA, responded to the Company’s Pre-RFD regarding product classification and jurisdictional assessment. The FDA’s OCP determined that VergenixSTR should be classified as a Combination Product, specifically a drug/biologic/device product, and should be assigned to the FDA’s CBER. A Pre-RFD is FDA’s preliminary, nonbinding assessment of (1) the regulatory identity or classification of a product as a drug, device, biological product, or combination product, and (2) which FDA Center (i.e., CBER, CDER, or CDRH) will have primary jurisdiction for the premarket review and regulation of the product. Therefore, this classification and jurisdictional assessment is subject to change. We currently do not intend to pursue an FDA regulatory pathway to market for VergenixSTR in 2019. We still include a discussion of FDA’s requirements for approval of and ongoing regulation for drugs, biologics, and medical devices below.
Marketing Authorization for Drugs and Biologics in the U.S.
A new biologic must be approved by the FDA through the biologics license application, or BLA, process before it may be legally marketed in the U.S. A new drug must be approved by the FDA through the new drug application, or NDA, process before it may be legally marketed in the U.S.
The animal and other non-clinical data and the results of human clinical trials performed under an Investigational New Drug, or IND, application and under similar foreign applications will become part of the BLA or NDA.
67
In the U.S., the FDA regulates biologics under the Public Health Service Act, or PHS Act, and implementing regulations, and under the Federal Food, Drug, and Cosmetic Act, or FDCA, and implementing regulations, respectively. The U.S. regulates drugs under the FDCA. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval may subject an applicant to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, requesting product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement, or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us. The process required by the FDA before a drug or biologic may be marketed in the U.S. generally involves the following:
| ● | completion of preclinical laboratory tests, animal studies and formulation studies according to Good Laboratory Practices, or GLP, or other applicable regulations; |
| ● | submission to the FDA of an IND which must become effective before human clinical trials may begin; |
| ● | approval by an institutional review board, or IRB, representing each clinical trial site before each clinical trial may be initiated; |
| ● | performance of adequate and well-controlled human clinical trials according to Good Clinical Practices, or GCP, to establish the safety and efficacy of the proposed biologic for its intended use; |
| ● | preparation and submission of a BLA or NDA to the FDA; |
| ● | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug is produced to assess compliance with current good manufacturing practice, or cGMP, to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and satisfactory completion of any FDA audits of the clinical study sites to assure compliance with GCP, and the integrity of clinical data in support of the BLA or NDA; and |
| ● | FDA review and approval of the BLA or NDA. |
Once a biologic or drug product candidate is identified for development, it enters the preclinical testing stage. Preclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information and analytical data, to the FDA as part of the IND. The sponsor will also include a protocol detailing, among other things, the objectives of the first phase of the clinical trials, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated if the first phase lends itself to an efficacy evaluation. Some preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, places the clinical trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Clinical holds also may be imposed by the FDA at any time before or during studies due to safety concerns or non-compliance.
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with GCP regulations. They must be conducted under protocols detailing the objectives of the trial, dosing procedures, subject selection and exclusion criteria, and the safety and effectiveness criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND, and progress reports detailing the results of the clinical trials must be submitted at least annually. In addition, timely safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events. An institutional review board, or IRB, responsible for the research conducted at each institution participating in the clinical trial must review and approve each protocol before a clinical trial commences at that institution and must also approve the information regarding the trial and the consent form that must be provided to each trial subject or his or her legal representative, monitor the study until completed and otherwise comply with IRB regulations.
| ● | Phase I: The product candidate is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, such as cancer, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing may be conducted in patients. |
68
| ● | Phase II: This phase involves studies in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
| ● | Phase III: Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical study sites. These studies are intended to establish the overall risk-benefit ratio of the product candidate and provide, if appropriate, an adequate basis for product labeling. |
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of a biologic or drug and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate, and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life. Before approving a BLA or NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in full compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. The PHS Act in particular emphasizes the importance of manufacturing control for products like biologics whose attributes cannot be precisely defined.
Manufacturers and others involved in the manufacture and distribution of products must also register their establishments with the FDA and certain state agencies. Both domestic and foreign manufacturing establishments must register and provide additional information to the FDA upon their initial participation in the manufacturing process. Any product manufactured by or imported from a facility that has not registered, whether foreign or domestic, is deemed misbranded under the FDCA.
Establishments may be subject to periodic unannounced inspections by government authorities to ensure compliance with cGMP and other laws. Manufacturers may have to provide, on request, electronic or physical records regarding their establishments. Delaying, denying, limiting, or refusing inspection by the FDA may lead to a product being deemed to be adulterated. Human clinical trials for biologics and drugs are typically conducted in three sequential phases that may overlap or be combined. If there are two independent modes of action, neither of which is subordinate to the other, the FDA makes a determination as to which center to assign the product based on consistency with other combination products raising similar types of safety and effectiveness questions or to the center with the most expertise in evaluating the most significant safety and effectiveness questions raised by the combination product.
Marketing Authorization for Medical Devices in the U.S.
In the United States, medical devices are regulated by the FDA. Unless an exemption applies, a new medical device will require either prior 510(k) clearance or approval of a PMA before it can be marketed in the United States. The information that must be submitted to the FDA in order to obtain clearance or approval to market a new medical device varies depending on how the medical device is classified by the FDA. Medical devices are classified into one of three classes on the basis of the controls deemed by the FDA to be necessary to reasonably ensure their safety and effectiveness. Class I devices, which are those that have the lowest level or risk associated with them, are subject to general controls, including labeling, premarket notification, and adherence to the QSR. Class II devices are subject to general controls and special controls, including performance standards. Class III devices, which have the highest level of risk associated with them, are subject to most of the previously identified requirements as well as to premarket approval. Most Class I devices and some Class II devices are exempt from the 510(k) requirement, although manufacturers of these devices are still subject to registration, listing, labeling and QSR requirements.
69
A 510(k) premarket notification must demonstrate that the device in question is substantially equivalent to another legally marketed device, or predicate device, that did not require premarket approval. In evaluating the 510(k), the FDA will determine whether the device has the same intended use as the predicate device, and: (i)(a) has the same technological characteristics as the predicate device, or (b) has different technological characteristics; and (ii)(a) the data supporting the substantial equivalence contains information, including appropriate clinical or scientific data, if deemed necessary by the FDA, that demonstrates that the device is as safe and as effective as a legally marketed device, and (b) does not raise different questions of safety and effectiveness than the predicate device. Most 510(k)s do not require clinical data for clearance, but the FDA may request such data. If the FDA does not agree that the new device is substantially equivalent to the predicate device, the new device will be classified in Class III, and the manufacturer must submit a PMA.
The PMA process is more complex, costly, and time consuming than the 510(k) clearance procedure. A PMA must be supported by extensive data including, but not limited to, technical, preclinical, clinical, manufacturing, control, and labeling information to demonstrate to the FDA’s satisfaction the safety and effectiveness of the device for its intended use. After a PMA is submitted, the FDA has 45 days to determine whether it is sufficiently complete to permit a substantive review. If the PMA is complete, the FDA will file the PMA. The FDA is subject to performance goal review times for PMAs and may issue a decision letter as a first action on a PMA within 180 days of filing, but if it has questions, it will likely issue a first major deficiency letter within 150 days of filing. It may also refer the PMA to an FDA advisory panel for additional review and will conduct a preapproval inspection of the manufacturing facility to ensure compliance with the QSR, either of which could extend the 180-day response target. A PMA can take several years to complete, and there is no assurance that any submitted PMA will ever be approved. Even when approved, the FDA may limit the indication for which the medical device may be marketed. Changes to the device, including changes to its manufacturing process, may require the approval of a supplemental PMA.
If a medical device is determined to present a “significant risk,” the manufacturer may not begin a clinical trial until it submits an investigational device exemption, or IDE, to the FDA and obtains approval of the IDE from the FDA. The IDE must be supported by appropriate data, such as animal and laboratory testing results, and include a proposed clinical protocol. The clinical trials must be conducted in accordance with applicable regulations, including but not limited to the FDA’s IDE regulations and current good clinical practices. A clinical trial may be suspended by the FDA or the sponsor at any time for various reasons, including a belief that the risks to the study participants outweigh the benefits of participation in the trial. Even if a clinical trial is completed, the results may not demonstrate the safety and efficacy of a device or may be equivocal or otherwise not be sufficient to obtain approval. Medical devices, however, typically rely on one or a few pivotal studies rather than Phase I, II and III clinical trials
Clinical trials are subject to extensive monitoring, recordkeeping and reporting requirements. Clinical trials must be conducted under the oversight of an institutional review board, or IRB, for the relevant clinical trial sites and must comply with FDA regulations, including, but not limited to, those relating to good clinical practices. To conduct a clinical trial, we also are required to obtain the patient’s informed consent in a form and substance that complies with both FDA requirements and state and federal privacy and human subject protection regulations.
The FDA, the IRB, or we could suspend a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the anticipated benefits or a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. Clinical testing may not be completed successfully within any specified period, if at all. Even if a trial is completed, the results of clinical testing may not adequately demonstrate the safety and efficacy of the device or may otherwise not be sufficient to obtain FDA clearance or approval to market the product in the United States. Similarly, in Europe, the clinical study must be approved by a local ethics committee and in some cases, including studies with high-risk devices, by the ministry of health in the applicable country.
In August 2010, we submitted a 510(k) notification to the FDA for VergenixWD, a collagen-based non-invasive dressing. In October 2010, we received notice that the Center for Devices and Radiological Health, or CDRH, which is the FDA center with jurisdiction over medical devices, determined that the product required a submission of a PMA for regulatory approval and not a 510(k). We filed an appeal of this decision which was denied, and in April 2012, the FDA confirmed its previous determination that our product would require PMA approval prior to its marketing in the United States. Most dermal fillers have been traditionally regulated as medical devices. However, similar products have more recently been regulated as biologics by CBER. Therefore, the classification and jurisdictional assessment related to our VergenixWD product is subject to change. We believe that most, if not all, of our products will be subject to the PMA process or will be considered combination products subject to at least some medical device regulations.
70
We expect, based on our prior limited interaction with the FDA in connection with our predecessor wound healing product, that our current products will be regulated as medical devices through a PMA process; however, no assurance can be given that the FDA will not impose additional, more stringent, regulatory requirements with respect to one or more of our current or future product candidates. Conducting clinical trials for our pipeline product candidates that are required to undergo the PMA process may take one to three years, depending on the composition of the product candidate under development and its designation.
We are not presently conducting any discussions with the FDA with respect to any of our products.
Post-Approval Regulation of Biologics, Drugs and Medical Devices
After a product is placed on the market, numerous regulatory requirements continue to apply. In addition to the requirements below, adverse event reporting regulations require that we report to the FDA any incident in which our product may have caused or contributed to a death or serious injury or in which our product malfunctioned and, if the malfunction were to recur, would likely cause or contribute to death or serious injury. Additional regulatory requirements include:
| ● | product listing and establishment registration, which helps facilitate FDA inspections and other regulatory action; |
| ● | cGMP or QSR, which requires manufacturers, including third-party manufacturers, to follow stringent design, validation, testing, control, documentation and other quality assurance procedures during all aspects of the design and manufacturing process; |
| ● | labeling regulations and FDA prohibitions against the promotion of products for uncleared, unapproved or off-label use or indication; |
| ● | clearance of product modifications that could significantly affect safety or effectiveness or that would constitute a major change in intended use of one of our approved medical products; |
| ● | notice or approval of product or manufacturing process modifications or deviations that affect the safety or effectiveness of one of our approved medical products; |
| ● | post-approval restrictions or conditions, including post-approval study commitments; |
| ● | post-market surveillance regulations, which apply, when necessary, to protect the public health or to provide additional safety and effectiveness data for the medical product; |
| ● | the FDA’s recall authority, whereby it can ask or, under certain conditions, order device manufacturers to recall from the market a product that is in violation of governing laws and regulations; |
| ● | regulations pertaining to voluntary recalls; and |
| ● | notices of corrections or removals. |
Also, quality control and manufacturing procedures must continue to conform to current Good Manufacturing Practices, or cGMP after approval, which includes, among other things, maintenance of a stability program. The FDA periodically inspects manufacturing facilities to assess compliance with cGMP, which imposes extensive procedural, substantive, and record keeping requirements. In addition, changes to the manufacturing process are strictly regulated and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of product out of specification results and impose reporting and documentation requirements upon us and any third-party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance. The holder of an NDA is responsible for legal and regulatory compliance for advertising and promotion of the drug product. We are required to provide to the FDA copies of all drug promotion at the time of first use and to ensure that all information disseminated conforms to the product’s approved labeling and other FDA regulations and policies.
71
A biologic product may also be subject to official lot release, meaning that the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official lot release, the manufacturer must submit samples of each lot, together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot, to the FDA. The FDA may, in addition, perform certain confirmatory tests on lots of some products before releasing the lots for distribution. Finally, the FDA will conduct laboratory research related to the safety, purity, potency and effectiveness of pharmaceutical products.
Advertising and promotion of medical devices, in addition to being regulated by the FDA, are also regulated by the U.S. Federal Trade Commission, or FTC, and by state regulatory and enforcement authorities. Promotional activities for FDA-regulated products of other companies have been the subject of enforcement action brought under healthcare reimbursement laws and consumer protection statutes. Furthermore, under the federal U.S. Lanham Act and similar state laws, competitors and others can initiate litigation relating to advertising claims. In addition, we are required to meet regulatory requirements in countries outside the United States, which can change rapidly with relatively short notice. If the FDA determines that our promotional materials or training constitutes promotion of an unapproved or uncleared use, it could request that we modify our training or promotional materials or subject us to regulatory or enforcement actions. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our promotional or training materials to constitute promotion of an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement.
Failure by us or by our third-party manufacturers and suppliers to comply with applicable regulatory requirements can result in enforcement action by the FDA or other regulatory authorities, which may result in sanctions including, but not limited to:
| ● | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; |
| ● | customer notifications or repair, replacement, refunds, recall, detention or seizure of our products; |
| ● | operating restrictions or partial suspension or total shutdown of production; |
| ● | refusing or delaying requests for 510(k) clearance or PMA approvals of new products or modified products; |
| ● | withdrawing 510(k) clearances or PMA approvals that have already been granted; |
| ● | refusing to grant export approval for our products; or |
| ● | criminal prosecution. |
Proteins Intended for Therapeutic Use
In the United States, proteins intended for therapeutic use, whether derived from plants, animals, microorganisms, or recombinant versions of these products, are regulated as biological products that have been transferred from the FDA Center for Biologics Evaluation and Research, or CBER, to the Center for Drug Evaluation and Research, or CDER. CDER has regulatory responsibility, including premarket review and continuing oversight over the transferred products. Cellular products, including products composed of human, bacterial, or animal cells, or from physical parts of those cells, remain under the jurisdiction of CDER.
72
Our products are based on our recombinant type I human collagen, or rhCollagen, a form of human collagen produced with our proprietary plant based genetic engineering technology. Therefore, we believe our underlying platform technology would be regulated as a drug in the U.S.
Regenerative Medicine Advanced Therapy Designation
Under section 3033 of the 21st Cures Act, or Cures Act, a drug is eligible for regenerative medicine advanced therapy (RMAT) designation if (1) the drug is a regenerative medicine therapy, which is defined as a cell therapy, therapeutic tissue engineering product, human cell and tissue product, or any Combination Product using such therapies or products, except for those regulated solely under section 361 of the PHS Act and 21 C.F.R. Part 1271, (2) the drug is intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition, and (3) preliminary clinical evidence indicates that the drug has the potential to address unmet medical needs for such disease or condition. If we pursue U.S. marketing approval for any of our products, we may be able to avail ourselves of this pathway or another expedited pathway.
Human Cells, Tissues, and Cellular and Tissue-Based Products Regulation
Under Section 361 of the PHS Act, the FDA issued specific regulations governing the use of human cells, tissues, and cellular and tissue-based products, or HCT/Ps, in humans. Pursuant to Part 1271 of Title 21 of the Code of Federal Regulations, or Part 1271, the FDA established a unified registration and listing system for establishments that manufacture and process HCT/Ps. The regulations also include provisions pertaining to donor eligibility determinations; current good tissue practices covering all stages of production, including harvesting, processing, manufacture, storage, labeling, packaging, and distribution; and other procedures to prevent the introduction, transmission, and spread of communicable diseases.
The HCT/P regulations strictly constrain the types of products that may be regulated solely under these regulations. Factors considered include the degree of manipulation, whether the product is intended for a homologous function, whether the product has been combined with noncellular or non-tissue components, and the product’s effect or dependence on the body’s metabolic function. In those instances where cells, tissues, and cellular and tissue-based products have been only minimally manipulated, are intended strictly for homologous use, have not been combined with noncellular or nontissue substances, and do not depend on or have any effect on the body’s metabolism, the manufacturer is only required to register with the FDA, submit a list of manufactured products, and adopt and implement procedures for the control of communicable diseases. If one or more of the above factors has been exceeded, the product would be regulated as a drug, biological product, or medical device rather than an HCT/P.
We do not believe that Part 1271 requirements currently apply to us because we are not currently investigating, marketing or selling cellular therapy products in the U.S. If we were to change our business operations in the future, the FDA requirements that apply to us may also change, and we would potentially need to expend significant resources to comply with these requirements.
European Union
Legal Requirements for Medical Devices in the EU
Under the European Union Medical Device Directive, or EU MDD, medical devices must meet the EU MDD requirements and receive a CE marking certification prior to marketing in the European Union, or EU. CE marking is the uniform labeling system of products designed to facilitate the supervision and control of the EU concerning manufacturers’ compliance with the various regulations and directives of the EU and to clarify the obligations imposed in the various legislative provisions in the EU. Use of a uniform product labeling indicates compliance with all of the directives and regulations required for the application of such labeling, and it is effective as a manufacturer’s declaration that the product meets the required criteria and technical specifications of the relevant authorities such as health, safety, and environmental protection. CE marking ensures free trade between the EU and European Economic Area (or EEA) countries (Switzerland, Iceland, Liechtenstein, and Norway) and permits the enforcement and customs authorities in European countries not to allow the marketing of similar products that do not bear the CE marking sign. Such certification allows, among other things, marking the products (according to various categories) with the CE marking and their sale and marketing in the EU.
73
CE marking certification under the MDD requires the performance of a conformity assessment procedure to establish that a product meets the essential requirements under the MDD. The nature of the conformity assessment procedure and the data required under it - including the question whether or not a clinical investigation of a device is required - depends on the risk class of the respective device. Devices of the lowest risk class, class I, are subject to self-certification by the manufacturer, while devices of higher risk classes, i.e., classes IIa, IIb and III, require a comprehensive quality system program, comprehensive technical documentation and data on the product, which are then reviewed by a Notified Body, or NB. An NB is an organization designated by the national governments of the EU member states to make independent judgments about whether a product complies with the EU MDD requirements and to grant the CE certificate if we, and our product, comply with specified terms. After receiving the CE certificate, we must pass a review carried out by the competent NB annually, under which it audits our facilities to verify our compliance with the ISO 13485 quality system standard. The CE-certificate is a requirement for the declaration of conformity we issue for our medical devices and for our legitimate affixing of the CE-mark to our products.
Compliance with the ISO 13485 standard, for medical device quality management systems, is required for regulatory purposes in the EU with regard to devices of risk class IIa or higher. ISO standards are recognized international quality standards that are designed to ensure that we develop and manufacture quality medical devices. Other countries are also instituting regulations regarding medical devices. Compliance with these regulations requires extensive documentation and clinical reports for all of our products, revisions to labeling, and other requirements such as facility inspections to comply with the registration requirements.
In February 2016, we received the CE certification for VergenixFG, and in October 2016, we received the CE certification for VergenixSTR. In December 2012, we received CE certification for VergenixWD in Europe. VergenixWD was our first medical product based on collagen protein derived from plants that is authorized for sale and marketing in Europe, but we are not currently promoting a marketing strategy for VergenixWD, which is considered a commodity product and is not targeted towards the advanced wound care market, which is our target market.
In February 2019, we received DEKRA (European Union Notified Body) Certification for the manufacturing and purification of our recombinant human collagen, rhCollagen. The Rehovot production facility is now covered by the current CollPlant ISO13485:2016 certification.
On May 27, 2020, the new Regulation Reg. EU No. 745/2017 on Medical Devices (Medical Devices Regulation) will become applicable and replace the existing regulatory framework for medical devices in the EU. The Medical Devices Regulation strengthens the medical devices rules in the EU. In particular, the Medical Devices Regulation will result in several medical devices being classified in higher risk classes and therefore face elevated regulatory requirements. In addition, the Medical Devices Regulation will generally elevate regulatory requirements to medical devices. As a result, it is likely that it will become more difficult to market medical devices and costs incurred for clinical evaluation, conformity assessment and post marketing surveillance will increase. These regulatory changes may adversely affect our business, financial condition, and results of operations or restrict our operations.
Legal Requirements for Drugs in the EU
We do not believe that our products are currently subject to EU or Member States’ regulation on drugs. However, given that our products are highly innovative, a risk remains that regulatory authorities, notified bodies, competitors and/or courts might be of a different opinion. Consequently, there is a risk that discussions might be started with regard to the regulatory status of our products.
If one or more of our current or future products would have the status of a drug under the law of the EU or one or more of its Member States, regulatory requirements for such product(s) would be significantly higher. In particular, a drug can only be placed on the market if it has been authorized by the competent regulatory authority either under the EU centralized procedure, the decentralized or mutual recognition procedure or under a Member State’s national procedure. Marketing authorizations for drugs under all of the different authorization procedures are expensive and time consuming and require the performance of extensive pre-clinical and clinical research. If one or more of our products would be considered drugs by a regulatory authority, notified body or court of the EU or a Member State, it is possible that we would be forced to take the respective product(s) off the market until they have received marketing approval under pharmaceutical law. In addition, this might also lead to administrative fines, criminal prosecution and/or claims raised by customers and/or competitors.
74
China
China’s medical device market, currently in a rapid state of expansion, is overseen by the National Medical Product Administration, or NMPA (formerly the China Food and Drug Administration). The NMPA issues registration certificates required for all medical devices sold in China. The NMPA uses a risk-based system, and its approval process requires mandatory testing for Class II and III devices. Class II devices are moderate-risk devices and Class III devices are high-risk medical devices. Third-party reviews of devices are currently not allowed in China; only the NMPA is authorized to approve devices. The registration process requires the submission of a registration standard along with device samples for testing. Manufacturers of Class II and Class III medical devices are also required to demonstrate that the device has been approved by the country of origin with documents like a CE certificate, 510(k) letter and PMA approval and compliance with ISO 13485, and they may also be required to submit clinical data in support of their application. In addition to these requirements, all medical device manufacturers must also include product information in Chinese on all packaging and labeling. Manufacturers exporting medical devices to China must appoint several China-based agents to act on their behalf. These include a registration agent to coordinate the NMPA registration process, a legal agent to handle any adverse events reported with a registered device, including a product recall, and an after-sales agent to provide technical service and maintenance support.
Other U.S. Federal Healthcare Laws and Regulations
Healthcare providers, physicians, and third-party payors play a primary role in the recommendation and medical devices that are granted marketing approval. In the United States, we are subject to laws and regulations pertaining to healthcare fraud and abuse, including anti-kickback laws and physician self-referral laws that regulate the means by which companies in the healthcare industry may market their products to hospitals and healthcare providers and may compete by discounting the prices of their products. The delivery of our products is subject to regulation regarding reimbursement, and federal healthcare laws apply when a customer submits a claim for a product that is reimbursed under a federally funded healthcare program. These rules require that we exercise care in structuring our sales and marketing practices and customer discount arrangements.
Arrangements with healthcare providers, third-party payors, and other customers are subject to broadly applicable fraud and abuse and other healthcare laws and regulations, including the following:
| ● | the federal healthcare Anti-Kickback Law prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving, or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order, or recommendation of, any good or service for which payment may be made, in whole or in part, under a federal healthcare program such as Medicare and Medicaid; |
| ● | the U.S. False Claims Act imposes civil penalties, and provides for civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease, or conceal an obligation to pay money to the federal government; |
| ● | the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters; |
| ● | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act and its implementing regulations, also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security, and transmission of individually identifiable health information; |
75
| ● | the federal false statements statute prohibits knowingly and willfully falsifying, concealing, or covering up a material fact or making any materially false statement in connection with the delivery of or payment for healthcare benefits, items, or services; |
| ● | the federal transparency requirements under the Health Care Reform Law require manufacturers of drugs, devices, and medical supplies to report to the U.S. Department of Health and Human Services information related to payments and other transfers of value to physicians and teaching hospitals and physician ownership and investment interests; and |
| ● | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental third-party payers, including private insurers. |
Healthcare providers that purchase medical devices generally rely on third-party payors, including, in the United States, the Medicare and Medicaid programs and private payors, such as indemnity insurers, employer group health insurance programs, and managed care plans, to reimburse all or part of the cost of the products. As a result, demand for our products is and will continue to be dependent in part on the coverage and reimbursement policies of these payors. The manner in which reimbursement is sought and obtained varies based upon the type of payor involved and the setting in which the product is furnished and utilized. Reimbursement from Medicare, Medicaid, and other third-party payors may be subject to periodic adjustments as a result of legislative, regulatory, and policy changes as well as budgetary pressures. Possible reductions in, or eliminations of, coverage or reimbursement by third-party payors, or denial of, or provision of uneconomical reimbursement for new products, may affect our customers’ revenue and ability to purchase our products. Any changes in the healthcare regulatory, payment, or enforcement landscape relative to our customers’ healthcare services has the potential to significantly affect our operations and revenue.
Other Approvals
Our international operations, as well as being an Israeli company, subject us to laws regarding sanctioned countries, entities, and persons; customs, import-export, and laws regarding transactions in foreign countries; and the U.S. Foreign Corrupt Practices Act and local anti-bribery and other laws regarding interactions with healthcare providers. Among other things, these laws restrict, and in some cases can prevent, United States companies from directly or indirectly selling goods, technology, or services to people or entities in certain countries. In addition, these laws require that we exercise care in structuring our sales and marketing practices in foreign countries.
In addition to the above regulations, we are and may be subject to regulation under country-specific federal and state laws, including, but not limited to, requirements regarding record keeping and the maintenance of personal information, including personal health information. As a public company whose securities will be registered pursuant to the Securities Act, we will be subject to U.S. securities laws and regulations, including the Sarbanes-Oxley Act. We also are subject to other present, and could be subject to possible future, local, state, federal, and non-U.S. regulations in countries in which we will distribute our products.
Israeli Ministry of Agriculture
The process of growth of transgenic plants and the treatment thereof is subject to the regulations published by the Israeli Ministry of Agriculture and the approval of the Ministry of Agriculture to engage in the cultivation of recombinant plants. Although the Ministry of Agriculture requirements do not necessarily apply to our operations, we hold a valid permit from the Plant Protection and Inspection Services Administration, or PPIS, for growing tobacco plants in greenhouses in the north of Israel, as well as in all of our subcontractors’ facilities.
Business Licensing
Under the Israeli Licensing of Businesses Law, to which our production site and laboratories are subject, operating a business without a license or temporary permit is a criminal offense. We have a business license for our laboratories and offices, in effect until December 31, 2019. We have a business license for our production site at Yessod Hama’ala, in effect until November 3, 2019.
76
Planning and Zoning
Our production sites and laboratories are subject to the Israeli Planning and Zoning Law, which sets provisions and obligations, inter alia, regarding the licensing process for a new building, including building permits, non-conforming use and easements, the supervision over its construction, and the required occupancy permits. According to the Planning and Zoning Law, work or use of land without a permit where such permit is required, a deviation from the permit granted, or use of agricultural land in violation of the law, constitutes a criminal offense.
Employees
As of December 31, 2018, we had 40 full-time employees, including ten in research and development, twenty-two in manufacturing and eight in sales, general and administrative positions. Seven of our employees have either MDs or PhDs. All of our employees are located in Israel. We believe our employee relations are good.
In addition, we employ a limited number of part-time employees on a temporary basis, as well as consultants and service providers.
Israeli labor laws govern the length of the workday, minimum wages for employees, procedures for hiring and dismissing employees, determination of the scope of severance pay, annual leave, sick days, advance notice of termination of employment, equal opportunity and anti-discrimination laws, and other conditions of employment. Subject to specified exceptions, Israeli law generally requires severance pay upon the retirement, death, or dismissal of an employee. We fund our ongoing severance obligations by making monthly payments to insurance policies that comply with the applicable Israeli legal requirements. All of our current employees have agreed that upon termination of their employment, they will be entitled to receive only the amounts accrued in the insurance policies with respect to severance pay. Furthermore, Israeli employers and employees are required to make payments to the National Insurance Institute, which is similar to the U.S. Social Security Administration.
None of our employees currently work under any collective bargaining agreements.
Environmental, Health, and Safety Matters
Our research, development, and manufacturing processes involve the controlled use of certain hazardous materials. Therefore, we are subject to extensive environmental, health, and safety laws and regulations in a number of jurisdictions in Israel, governing, among other things: the use, storage, registration, handling, emission, and disposal of chemicals, waste materials, and sewage; chemicals, air, water, and ground contamination; air emissions; and the cleanup of contaminated sites, including any contamination that results from spills due to our failure to properly dispose of chemicals, waste materials, and sewage. Our operations at our Ness-Ziona manufacturing facility use chemicals and produce waste materials and sewage. Our activities require permits from various governmental authorities including local municipal authorities, the Ministry of Environmental Protection, and the Ministry of Health. The Ministry of Environmental Protection, the Ministry of Health, local authorities, and the municipal water and sewage company conduct periodic inspections in order to review and ensure our compliance with various regulations.
These laws, regulations, and permits could potentially require the expenditure by us of significant amounts for compliance or remediation. We believe that our environmental, health, and safety procedures for handling and disposing of these materials comply with the standards prescribed by the controlling laws and regulations. If we fail to comply with such laws, regulations, or permits, we may be subject to fines and other civil, administrative, or criminal sanctions, including the revocation of permits and licenses necessary to continue our business activities. In addition, we may be required to pay damages or civil judgments with respect to third-party claims, including those relating to personal injury (including exposure to hazardous substances we use, store, handle, transport, manufacture, or dispose of), property damage, or contribution claims. These risks are managed to minimize or eliminate associated business impacts. Some environmental, health, and safety laws allow for strict joint and several liability for remediation costs, regardless of comparative fault. We may be identified as a responsible party under such laws. Such developments could have a material adverse effect on our business, financial condition, and results of operations as these kinds of liabilities could exceed our resources. We could be subject to a regulatory shutdown of a facility that could prevent the distribution and sale of products manufactured in such facility for a significant period of time, and we could suffer a casualty loss that could require a shutdown of the facility in order to repair it, any of which could have a material, adverse effect on our business. Although we continuously strive to maintain full compliance with respect to all applicable global environmental, health, and safety laws and regulations, we could incur substantial costs to fully comply with future laws and regulations, and our operations, business, or assets may be negatively affected.
77
In addition, compliance with laws and regulations relating to environmental, health, and safety matters is an ongoing process and is often subject to change. In the event of any changes or new laws or regulations, we could be subject to new compliance measures or to penalties for activities which were previously permitted. For instance, Israeli regulations were promulgated in 2012 relating to the discharge of industrial sewage into the sewer system. These regulations establish new and potentially significant fines for discharging forbidden or irregular sewage into the sewage system. We have compliance procedures in place for employee health and safety programs, driven by a centrally led organizational structure that ensures proper implementation, which is essential to our overall business objectives.
We invest resources in creating a green production environment and in the treatment and disposal of waste using environmentally friendly processes. We have received all the necessary permits from the Ministry of Environmental Protection regarding our operations in Yessod Hama’ala and Ness-Ziona. We consult with environmental consultants for direction on environmental issues.
Legal Proceedings
From time to time we may become involved in legal proceedings or be subject to claims arising in the ordinary course of our business. We are currently not a party to any material legal or administrative proceedings and, except as set forth below, are not aware of any pending or threatened material legal or administrative proceedings against us.
78
On September 6, 2017, we received a VAT assessment from the Israel Tax Authority according to which we are required to pay tax in the amount of NIS 1.5 million, plus linkage differentials and interest, for the years 2012-2016. We disputed the position of the Israel Tax Authority resulting the latter to increase its VAT assessment and require us to pay tax in the amount of NIS 1.8 million (including linkage differentials and interest) for the abovementioned period. We have appealed the entire assessment to the District Court, in view of our position that we are not liable for the entire tax requirement. A preliminary hearing in the District Court in Lod, Israel is scheduled for May 2019. Our position relies, among other things, on an agreement signed between us and the Israel Tax Authority in 2011, which allows us to deduct VAT as stated.
| C. | Organizational Structure |
We currently have one wholly owned subsidiary: CollPlant Ltd., which is incorporated in the State of Israel.
| D. | Property, Plant and Equipment |
From March 31, 2019, our corporate headquarters and research facilities are located in the Weizmann Science Park in Rehovot, Israel. In November 2018, we entered into a new lease for an aggregate of approximately 13,450 square feet of office and laboratory space. The rent period effectively began on March 31, 2019. The term of the lease is for 65 months, commencing on November 15, 2018 and ending on April 15, 2024, with an option to extend the lease for an additional five years. Monthly rent is NIS 89,000. We have invested, and expect to further invest approximately NIS 3.6 million in improvements in the infrastructure, offices, labs and equipment in our new office space, net of participation by the landlord. From June 2008 to March 31, 2019, we leased an aggregate of approximately 7,653 square feet of office and laboratory space in Ness-Ziona, pursuant to lease agreements which expired, following extensions, on March 31, 2019.
We rent additional areas in Yessod Hama’ala, Israel, of approximately 64,583 square feet of greenhouse and manufacturing facility pursuant to a lease agreement that expires on April 20, 2021. In addition, on July 28, 2016, we leased additional space in Rehovot, Israel, of approximately 6,329 square feet for development and production activities pursuant to a lease agreement that expires on July 28, 2019, with an option to extend for four additional years.
The majority of our research and development work is carried out at our offices and research laboratories in Weizmann Science Park, Israel. The plant research process of our rhCollagen is carried out at our site in the north Israel. We use greenhouses for tobacco growing and other development services in several areas in Israel, where we are using subcontractors under several agreements. We produce our rhCollagen and BioInk in our two production sites, in the north of Israel and in Rehovot.
79
We believe that our existing facilities are adequate for our near-term needs. When our leases expire, we may look for extension periods or alternate space for our operations. We believe that suitable additional or alternative space and area would be available if required in the future on commercially reasonable terms.
| ITEM 4A. | UNRESOLVED STAFF COMMENTS |
Not applicable.
| ITEM 5. | OPERATING AND FINANCIAL REVIEW AND PROSPECTS |
You should read the following discussion and analysis of our financial condition and results of operations together with the section titled “Item 3.A.—Selected Financial Data” and our consolidated financial statements and related notes included elsewhere in this Annual Report on Form 20-F. This discussion and other parts of this Annual Report on Form 20-F contain forward-looking statements based upon current expectations that involve risks and uncertainties. This discussion and other parts of this Annual Report on Form 20-F contain forward-looking statements that involve risk and uncertainties, such as statements of our plans, objectives, expectations, and intentions. Our actual results could differ materially from those discussed in these forward looking statements. Factors that could cause or contribute to such differences include, but are not limited to, those discussed in the section titled “Item 3.D.—Risk Factors” and elsewhere in this Annual Report in Form 20-F.
The share and per share numbers in the following discussion reflect a 1-for-3 reverse share split that we effected on November 20, 2016. We report financial information under IFRS as issued by the International Accounting Standards Board, and none of the financial statements were prepared in accordance with generally accepted accounting principles in the United States.
Overview
We are a regenerative medicine company focused on developing and commercializing tissue repair products, initially for 3D bioprinting of tissues and organs, dermal fillers for aesthetics, orthobiologics, and advanced wound care markets. Our products are based on our rhCollagen, a form of human collagen produced with our proprietary plant-based genetic engineering technology. We believe our technology is the only commercially viable technology available for the production of genetically engineered, or recombinant, human collagen. We believe that our rhCollagen, though laboratory-derived, is identical to the type I collagen produced by the human body and has significant advantages compared to currently available tissue-derived collagens, including improved biofunctionality, high homogeneity, and reduced risk of immune response. We believe the attributes of our rhCollagen make it suitable for numerous tissue repair applications throughout the human body. We believe that the annual market size for our BioInk, VergenixSTR, VergenixFG and our dermal filler exceeded $8 billion in 2016 and is estimated to reach $12 billion in 2024.
Our rhCollagen-based BioInk for use in the 3D printing of tissues and organs is being developed to enable the printing of three-dimensional scaffolds combined with human cells and/or growth factors as a basis for tissue or organ formation. In addition to collagen, our BioInk formulations can include other proteins and/or polymers as well. Our BioInk is being developed to be compatible with numerous 3D bioprinting technologies and with printed organ characteristics. In October 2018, we entered into the United License Agreement, pursuant to which CollPlant and LB will collaborate in the development of engineered lungs or lung substitutes using our rhCollagen and BioInk.
Our VergenixSTR product is a soft tissue repair matrix that combines cross-linked rhCollagen with platelet-rich plasma, or PRP, a concentrated blood plasma that contains high levels of platelets and, in the European Union, is intended for the treatment of tendinopathy. We commenced commercial sales of VergenixSTR in December 2016. Prior to that, in August 2016, we completed an open label, single arm, multi-center clinical trial of VergenixSTR of 40 patients in Israel to demonstrate safety and to evaluate the performance of VergenixSTR in patients suffering from tennis elbow or lateral epicondylitis, an inflammation of the tendons that join the forearm muscles on the outside of the elbow. In October 2016, we received CE marking for VergenixSTR, which is required for a product to be marketed in the European Union, and in November 2016, we entered into an exclusive distribution agreement with Arthrex for VergenixSTR covering Europe, the Middle East, India, and certain African countries. In November 2018, we entered into an exclusive distribution agreement with Joinsmart Biomedical Co. for VergenixSTR in Taiwan. The distributor is currently in the process of certification and authorization of VergenixSTR, as required in Taiwan.
80
Our VergenixFG product is a wound-filling flowable gel made from our rhCollagen, which in the European Union is intended for treatment of deep surgical incisions and wounds including diabetic ulcers, burns, bedsores, and other chronic wounds. We completed an open label, single arm, multi-center clinical trial of VergenixFG of 20 patients in Israel to demonstrate safety and to evaluate the performance of VergenixFG in patients with hard-to-heal chronic wounds of the lower limbs. In February 2016, we received CE marking certification for VergenixFG, and in July 2016, we supplied our first order in Europe.
To date, we have financed our operations primarily with the net proceeds from private placements and from public offerings of our securities on the TASE, participation in product development collaborations, and government grants from the IIA.
Since our inception, we have incurred significant losses. Our total comprehensive loss was NIS 13.9 million for the year ended December 31, 2018 and NIS 20.9 million and NIS 27.9 million for the years ended December 31, 2017 and 2016, respectively. As of December 31, 2018, we had an accumulated deficit of NIS 184.7 million.
We expect to continue to incur expenses and operating losses for the foreseeable future. The net losses we incur may fluctuate significantly from quarter to quarter. We anticipate that our expenses will increase substantially if and as we:
| ● | continue our research and preclinical and clinical development of our pipeline products; |
| ● | seek marketing approvals for VergenixSTR and VergenixFG and any other products in the United States and other new territories; |
| ● | maintain, expand, and protect our intellectual property portfolio; |
| ● | hire additional operational, clinical, quality control, and scientific personnel; |
| ● | establish plant infrastructure to accommodate product capacity increase; |
| ● | add operational, financial, and management information systems and personnel, including personnel to support our product development, any future commercialization efforts, and our transition to a public reporting company in the United States; and |
| ● | identify additional product candidates. |
Financial Operations Overview
Revenue
Our ability to generate significant revenues depend on the successful commercialization of our rhCollagen-based BioInk, VergenixSTR and VergenixFG. In the year ended December 31, 2018, we reported revenues of NIS 18.0 million ($4.8 million) from the United License Agreement and the sale of rhCollagen-based BioInk in the United States and from the sales of VergenixSTR and VergenixFG in Europe.
Our revenues are measured at fair value of the consideration received or receivable for the sale of goods in the ordinary course of business. Revenues are recognized to the extent that it is probable that the economic benefits will flow to us and the revenues can be reliably measured. Revenues from the sale of products are recognized when all the significant risks and rewards of ownership of the products have passed to the buyer.
81
Operating Expenses
Research and Development Expenses
Research and development expenses consist of costs incurred for the development of both of our rhCollagen based developed products. Those expenses include:
| ● | employee-related expenses, including salaries and share-based compensation expenses for employees in research and development functions; |
| ● | expenses incurred in operating our laboratories; |
| ● | expenses incurred under agreements with CROs and investigative sites that conduct our clinical trials; |
| ● | expenses relating to outsourced and contracted services, such as external laboratories, consulting, and advisory services; |
| ● | supply, development, and manufacturing costs relating to clinical trial materials; |
| ● | maintenance of facilities, depreciation, and other expenses, which include direct and allocated expenses for rent and insurance, net of expenses capitalized to inventory; and |
| ● | costs associated with preclinical and clinical activities. |
Research and development activities are the primary focus of our business. Products in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of later-stage clinical trials. We expect that our research and development expenses will continue to be significant in absolute dollars in future periods as we continue to invest in research and development activities related to the development of our products.
Our total research and development expenses for the years ended December 31, 2018, December 31, 2017 and December 31, 2016 were NIS 16.2 million ($4.3 million), NIS 16.9 million ($4.9 million) and NIS 29.2 million ($7.6 million), respectively. The research and development expenditures on our rhCollagen technology and our products for the year ended December 31, 2018 include royalties expenses to the IIA in the amounts of NIS 3.2 million ($861,000) net of government grants. In the years ended December 31, 2017 and December 31, 2016 these expenses were partly funded in the amounts NIS 2.9 million ($836,000) and NIS 12.4 million ($3.2 million), respectively, by Bioventus and government grants net of royalties expenses. To date we have charged all research and development expenses to operations as they are incurred.
There are numerous factors associated with the successful commercialization of any of our products, including future trial design and various regulatory requirements, many of which cannot be determined with accuracy at this time. Additionally, future commercial and regulatory factors beyond our control will affect our clinical development programs and plans.
Participation in Research and Development Expenses
Our research and development expenses are net of the following participations by third parties.
Participation by the Israel Innovation Authority. We have received grants from IIA part of the research and development programs for our rhCollagen technology and our products. The requirements and restrictions for such grants are found in the Innovation Law and the regulations promulgated thereunder. These grants are subject to repayment through future royalty payments on any products resulting from these research and development programs, including VergenixSTR and VergenixFG. Under the Innovation Law and related regulations, royalties of 3% - 6% on the income generated from sales of products and from related services developed in whole or in part under IIA programs are payable to the IIA, up to the total amount of grants received, linked to the U.S. dollar and bearing interest at an annual rate of LIBOR applicable to U.S. dollar deposits, as published on the first business day of each calendar year. The total gross amount of grants actually received by us from the IIA as of December 31, 2018 totaled approximately NIS 37.4 million ($10.0 million). As of December 31, 2018, we paid NIS 5.5 million ($1.5 million) to the IIA.
82
In addition to paying any royalty due, we must abide by other restrictions associated with receiving such grants under the Innovation Law that continue to apply following repayment to the IIA. These restrictions may impair our ability to outsource manufacturing or otherwise transfer our know-how outside of Israel and may require us to obtain the approval of the IIA for certain actions and transactions and pay additional royalties and other amounts to the IIA. For more information, see “Item 3.D. Risk Factors—Risks Related to Our Financial Condition and Capital Requirements—The IIA grants we have received for research and development expenditures restrict our ability to manufacture products and transfer know-how outside of Israel and require us to satisfy specified conditions.” If we fail to comply with the Innovation Law, we may be subject to civil claims and criminal charges.
Research and development grants received from the IIA are recognized upon receipt as a liability if future economic benefits are expected from the project that will result in royalty-bearing sales. The amount of the liability for the loan is first measured at fair value using a discount rate that reflects a market rate of interest that reflects the appropriate degree of risks inherent in our business. The change in the fair value of the liability associated with grants from the IIA is reflected as an increase or decrease in our research and development expenses for the relevant quarter.
Under applicable accounting rules, the grants from the IIA have been accounted for as an off-set against the related research and development expenses in our financial statements. Our balance sheet liabilities include obligations regarding royalties that we are obligated to pay to the IIA based on future sales of our products. As a result, our research and development expenses are shown on our financial statements net of the IIA grants, and the participation in research and development expenses are shown on our financial statements net of the provision for IIA royalties. See Note 2G in our consolidated financial statements for the year ended December 31, 2018 for more information.
Participation by collaborators. In 2011, we entered into a joint development agreement with Pfizer for the development of a product for the orthopedic market, the Surgical Matrix Carrier, comprised of a growth factor and our rhCollagen, along with other components. This agreement expired in 2013. From 2013 to 2017, this co-development continued with Bioventus, which acquired the rights for commercialization of the growth factor from Pfizer and to whom Pfizer assigned certain of its rights and obligations under the 2011 joint development agreement. In March 2017, the co-development with Bioventus terminated.
General, Administrative, and Marketing Expenses
Our general and administrative expenses consist principally of:
| ● | employee-related expenses, including salaries, benefits, and related expenses, including equity-based compensation expenses; |
| ● | legal and professional fees for auditors and other consulting expenses not related to research and development activities; |
| ● | cost of offices, communication, and office expenses; |
| ● | information technology expenses; and |
| ● | business development and marketing activities. |
We expect that our general, administrative, and marketing expenses will increase in the future as our business expands and we incur additional general and administrative costs associated with being a public company in the United States, including compliance under the Sarbanes-Oxley Act and rules promulgated by the SEC. These public company-related increases will likely include costs of additional personnel, additional legal fees, audit fees, directors’ liability insurance premiums, and costs related to investor relations. We also expect that our marketing expenses will increase, as we will incur additional marketing costs associated with the commencement of sales, when and if our products are approved.
83
Financial Income/Financial Expense
Financial income includes interest income regarding short term deposits and exchange rate differences. Financial expense consists primarily of exchange rate differences and bank commissions.
Taxes on Income
We do not generate taxable income in Israel, as we have historically incurred operating losses resulting in carry forward tax losses. As of December 31, 2018, we have incurred operating losses of approximately NIS 152 million ($41 million) for CollPlant Holdings Ltd. and NIS 12.6 million ($3.4 million) for CollPlant Ltd. We anticipate that we will be able to carry forward these tax losses indefinitely to future tax years assuming that we utilize them at the first opportunity. Accordingly, we do not expect to pay taxes in Israel until we have taxable income after the full utilization of our carry forward tax losses.
The standard corporate tax rate in Israel is 23%. Under the Investment Law, and other Israeli laws, we may be entitled to certain additional tax benefits, including reduced tax rates, accelerated depreciation, and amortization rates for tax purposes on certain assets and amortization of other intangible property rights for tax purposes.
| A. | Operating Results |
The table below provides our results of operations for the years ended December 31, 2018, 2017 and 2016.
| Year ended December 31, | ||||||||||||||||
| 2016 | 2017 | 2018 | 2018 | |||||||||||||
| (NIS in thousands) | (Convenience translation into USD in thousands(1)) |
|||||||||||||||
| Statement of comprehensive loss data: | ||||||||||||||||
| Revenue | 292 | 1,668 | 18,034 | 4,812 | ||||||||||||
| Cost of revenue | — | 52 | 1,426 | 380 | ||||||||||||
| Gross Profit | 292 | 1,616 | 16,608 | 4,432 | ||||||||||||
| Research and development expenses | 29,200 | 16,921 | 16,213 | 4,325 | ||||||||||||
| Participation in research and development expenses | (12,411 | ) | (2,855 | ) | 3,227 | 861 | ||||||||||
| Research and development expenses, net | 16,789 | 14,066 | 19,440 | 5,186 | ||||||||||||
| General, administrative, and marketing expenses | 11,048 | 8,303 | 12,480 | 3,330 | ||||||||||||
| Operating loss | (27,545 | ) | (20,753 | ) | (15,312 | ) | (4,084 | ) | ||||||||
| Financial income | 93 | 253 | 1,650 | 440 | ||||||||||||
| Financial expenses | (441 | ) | (380 | ) | (225 | ) | (60 | ) | ||||||||
| Financial (expenses) income, net | (348 | ) | (127 | ) | 1,425 | 380 | ||||||||||
| Comprehensive loss | 27,893 | 20,880 | 13,887 | 3,704 | ||||||||||||
| (1) | Calculated using the exchange rate reported by the Bank of Israel for December 31, 2018 at the rate of one U.S. dollar per NIS 3.748. |
Revenues
We generated revenues from the sale of VergenixFG, VergenixSTR, and our BioInk, as well as revenues from the United License Agreement in the year ended December 31, 2018 of approximately NIS 18.0 million ($4.8 million) compared to NIS 1.7 million ($481,000) in the year ended December 31, 2017. The increase in revenues in 2018 was primarily due to revenues recognized from the United License Agreement in the amount of NIS 14.7 million ($4.0 million) which comprised of NIS 11.0 million ($3.0 million) from the license and NIS 3.7 million ($1.0 million) from proceeds regarding the payment to the IIA. In addition, we had an increase in revenues from product sales and rendering of services in the total amount of NIS 1.6 million ($427,000).
84
We generated revenues from the sale of VergenixFG, VergenixSTR, and our BioInk in the year ended December 31, 2017 of approximately NIS 1.7 million ($481,000) compared to NIS 292,000 ($76,000) in the year ended December 31, 2016. The increase in sales in 2017 was due to initial sales of BioInk in the amount of NIS 689,000 ($199,000) and an increase in the volume of VergenixFG and VergenixSTR sales activity which was due to a number of factors including the following: (i) sales for the year ended December 31, 2017 included twelve months of sales activity of VergenixFG and VergenixSTR while the comparable period only included less than six months for VergenixFG as the first commercial sales commenced in July 2016, and less than one month for VergenixSTR as the first commercial sales commenced in December 2016, and (ii) the number of European territories has expanded from two territories for Vergenix FG and Vergenix STR combined in the year ended December 31, 2016 to more than ten territories for VergenixFG and VergenixSTR combined in the year ended December 31, 2017. As the demand for our products has increased, there was no material change in the average product’s sale prices.
Cost of revenue
We incurred cost of revenue in the amount of NIS 1.4 million ($380,000) in the year ended December 31, 2018 compared to NIS 52,000 ($15,000) in the year ended December 31, 2017. Cost of revenue includes mainly the cost of VergenixFG, VergenixSTR, and our rhCollagen based BioInk. The increase in cost of revenue in the amount of NIS 1.3 million ($367,000) is primarily due to the increase in revenues generated from BioInk sales.
Research and Development Expenses
We incurred research and development expenses of NIS 16.2 million ($4.3 million) in the year ended December 31, 2018 compared to NIS 16.9 million ($4.9 million) in the year ended December 31, 2017. The expenses primarily related to the development of our BioInk product, other development projects such as our aesthetics dermal filler and process development in our Rehovot facility. The decrease in the amount of NIS 700,000 ($187,000) is mainly due to reduction in research and development subcontractors expenses.
The participation by parties not affiliated with the Company in the research and development expenses are presented at net and accumulates to NIS 3.2 million ($861,000) in 2018, compared to NIS 2.9 million ($836,000) in 2017. The gross participation in 2018 accumulates to NIS 1.3 million ($347,000) before deduction of royalties’ payments to the IIA in connection with the United License Agreement, in the amounts of NIS 4.7 million ($1.2 million) and income from re-measurement of the liability to the IIA. Excluding the royalties payments, the decrease was in the amount of NIS 1.7 million ($454,000), and is mainly due to a decrease in income from re-measurement of the liability to the IIA amounting to NIS 832,000 ($222,000) and a decrease from termination of the development of the Surgical Matrix Carrier in March 2017, a project that was fully funded by Bioventus amounting to NIS 537,000 (143,000).
Research and development expenses decreased from NIS 16.9 million ($4.9 million) in the year ended December 31, 2017 compared to NIS 29.2 million ($7.6 million) in the year ended December 31, 2016. The expenses primarily related to the development of our BioInk, VergenixSTR and VergenixFG, and other development projects. In addition, expenses include development costs related to the Surgical Matrix Carrier incurred through the end of March 2017. The total decrease in expenses amounting to NIS 12.3 million ($3.5 million) is primarily due to (i) a decrease in subcontractors and consumables expenses attributable to a reduction in the amount of NIS 8.0 million ($2.3 million) in production and product development cost and expenses in the amount of NIS 1.8 ($519,000) million related to the Surgical Matrix Carrier, a project that ended in March 2017, and (ii) a reduction in salaries and amortization of equity-based compensation in the amount of NIS 2.0 million ($577,000) related to a reduction of development staff costs.
The participation by parties not affiliated with the Company in the research and development expenses was NIS 2.9 million ($836,000) in 2017, compared to NIS 12.4 million ($3.2 million) in 2016. The decrease in the amount of NIS 9.5 million ($2.4 million) is mainly due to termination of the development of the Surgical Matrix Carrier in March 2017, a project that was fully funded by Bioventus.
General, Administrative, and Marketing Expenses
We incurred general, administrative, and marketing expenses of NIS 12.5 million ($3.3 million) in the year ended December 31, 2018, compared to NIS 8.3 million ($2.4 million) in the year ended December 31, 2017. The total increase in expenses amounting to NIS 4.2 million ($1.1 million) is primarily due to (i) an increase in legal and professional expenses of NIS 1.6 million ($427,000) related to our US capital market expenses as a Nasdaq traded company, and (ii) an increase in salaries and amortization of equity-based compensation in the amount of NIS 2.6 million ($694,000).
85
We incurred general, administrative, and marketing expenses of NIS 8.3 million ($2.4 million) in the year ended December 31, 2017, compared to NIS 11.0 million ($2.9 million) in the year ended December 31, 2016. The decrease is primarily attributable to a decrease of NIS 2.7 million ($720,000) related to costs of our fundraising efforts in the U.S. in 2016.
Financial Expenses (Income), Net
Financial income, net, totaled NIS 1.4 million ($380,000) in the year ended December 31, 2018, compared to financial expense, net of NIS 127,000 ($36,000) in the year ended December 31, 2017. The increase in financial income in 2018 as compared to the year 2017 was mainly due to (i) remeasurement of financial instruments amounting to NIS 891,000 ($238,000), and (ii) exchange rate differences in the U.S. dollar exchange rate against the NIS amounting to NIS 759,000 ($203,000), where the U.S. dollar exchange rate increased compared to the NIS, and affected our U.S. dollar currency cash and cash equivalents.
Financial expenses, net, totaled NIS 127,000 ($36,000) in the year ended December 31, 2017, compared to financial expense, net of NIS 348,000 ($90,000) in the year ended December 31, 2016. The decrease in 2017 as compared to the same period in 2016 was due to exchange rate differences in the U.S. dollar exchange rate against the NIS, where the U.S. dollar exchange rate decreased compared to the NIS, and affected our U.S. dollar currency cash and cash equivalents and affected our liability for remaining obligations to the IIA.
Significant Accounting Estimates and Judgments
Estimates and judgments are reviewed on an ongoing basis and are based on past experience and other factors, including expectations of future events, which are considered reasonable in view of current circumstances.
Significant Accounting Estimates
We make estimates and assumptions with respect to the future. By nature, the accounting estimates are rarely identical to actual results. The estimate that has a significant risk of resulting in a material adjustment to carrying amounts of assets and liabilities in the next financial year is listed below.
Impairment of In Process Research and Development
We annually review the need to record impairment of in process research and development, or IPR&D. To test for impairment, we as a whole have been identified as the smallest cash-generating unit to which the intangible asset can be attributed. Accordingly, we measure our recoverable amount as a whole. The recoverable amount is the higher of value in use and fair value less costs of disposal. In accordance with IFRS 13, the quoted market price in an active market provides the most reliable evidence of fair value. Since fair value less costs of disposal, which is based on our market price, is significantly higher than the carrying amount of the cash-generating unit, we determined that no impairment exists.
Fair value measurement of debentures
We measured the fair value of the debentures based on accepted valuation models and assumptions regarding unobservable inputs used in the valuation models. See also Note 12 to the financial statements.
Significant Judgments Made When Applying our Accounting Policies
Grants from the IIA
In accordance with the accounting treatment prescribed in Note 2G to our financial statements appearing elsewhere in this Annual Report on Form 20-F, our management is required to examine whether there is reasonable assurance that the IIA grant that was received will be repaid. In addition, if, at the date of initial recognition, the grant is recognized in the statement of comprehensive income (loss), then in subsequent periods our management is required to evaluate whether it is no longer reasonably assured that royalties will not be paid to the IIA. In such a case, a liability would be recognized based on our best estimate of the amount required to settle our royalty obligation to the IIA.
86
As of December 31, 2018, grants received were recorded against the related research and development expenses in the statement of comprehensive loss.
As of December 31, 2018, two of our products for the orthobiologics and advanced wound care markets received marketing clearance in Europe. Following the signing of distribution agreements for more than ten different countries and the supply of orders for VergenixFG, and the distribution agreement signed with Arthrex for VergenixSTR, we believe that, as of December 31, 2018, there is reasonable assurance that NIS 1.2 million of royalties will be paid to the IIA and a liability is included in our financial statements as of December 31, 2018.
Development Costs
Development costs are capitalized in accordance with the accounting policy described in Note 2E(3) to our financial statements appearing elsewhere in this Annual Report on Form 20-F. Capitalization of costs is based on management’s judgment about technological and economic feasibility.
Our management believes that as of December 31, 2018, the above conditions were not met; therefore development costs were not capitalized.
Recent Accounting Pronouncements
IFRS 16 Leases
IFRS 16 will replace, upon first-time implementation, the existing guidance in IAS 17—Leases, or IAS 17. The standard sets out the principles for the recognition, measurement, presentation, and disclosure of leases, and is expected to have a material impact mainly on the accounting treatment applied by the lessee in a lease transaction.
IFRS 16 changes the existing guidance in IAS 17 and requires lessees to recognize a lease liability that reflects future lease payments and a “right-of-use asset” in all lease contracts (except for the following exemption), with no distinction between financing and capital leases. IFRS 16 exempts lessees in short-term leases or when the underlying asset has a low value.
IFRS 16 changes the definition of a “lease” and the manner of assessing whether a contract contains a lease.
IFRS 16 is effective retrospectively for annual periods beginning on or after January 1, 2019, taking into account the relief specified in the transitional provisions of IFRS 16. We are assessing the expected impact of IFRS 16 on our financial statements as described in Note 2T(1) to our financial statements.
JOBS Act
With less than $1.07 billion in revenues during our last fiscal year, we qualify as an emerging growth company under the JOBS Act. An emerging growth company may take advantage of specified provisions in the JOBS Act that provide exemptions or reductions of its regulatory burdens related to reporting and other requirements that are otherwise applicable generally to public companies. These provisions include an exemption from the auditor attestation requirement in the assessment of our internal control over financial reporting pursuant to the Sarbanes-Oxley Act. We may take advantage of some, but not necessarily all, of these provisions to reduce our burdens or exempt ourselves from regulatory requirements for up to five years or such earlier time that we are no longer deemed an emerging growth company. We have elected not to avail ourselves of an exemption that allows emerging growth companies to extend the transition period for complying with new or revised financial accounting standards. This election is irrevocable. We will be an emerging growth company until the earliest of: (i) the last day of the fiscal year during which we had total annual gross revenues of $1.07 billion or more, (ii) the last day of the fiscal year following the fifth anniversary of the date of the first sale of the ADSs pursuant to an effective registration statement, (iii) the date on which we have, during the previous three-year period, issued more than $1 billion in non-convertible debt, or (iv) the date on which we are deemed a “large accelerated filer” as defined in Regulation S-K under the Securities Act.
87
| B. | Liquidity and Capital Resources |
To date, we have financed our operations primarily with the net proceeds from private placements and from public offerings of our securities on the TASE, participation from product development collaborations, and government grants from the IIA.
Our recurring operating losses, negative cash flows and current cash position have raised substantial doubt regarding our ability to continue as a going concern. Our financial statements include a note describing the conditions which raise this substantial doubt. As a result, our independent registered public accounting firm included a “going concern” explanatory paragraph in its report on our financial statements as of and for the year ended December 31, 2018 with respect to this uncertainty. Our ability to continue as a going concern will require us to obtain additional financing to fund our operations. The perception of our ability to continue as a going concern may make it more difficult for us to obtain financing for the continuation of our operations and could result in the loss of confidence by investors, suppliers and employees. If we are not successful in raising capital through public or private offerings or reducing our expenses, we may exhaust our cash resources and will be unable to continue our operations. If we cannot continue as a viable entity, our shareholders would likely lose most or all of their investment in us.
We believe that, based on our current business plan, our existing cash and cash equivalents will be able to maintain our current planned development, manufacturing and marketing activities and the corresponding level of expenditures into the first quarter of 2020. This has led management to conclude in part that substantial doubt about our ability to continue as a going concern exists. In the event we are unable to successfully raise additional capital during or before the end of 2019, we will not have sufficient cash flows and liquidity to finance our business operations as currently contemplated. Accordingly, in such circumstances we would be compelled to immediately reduce general and administrative expenses and delay research and development projects and clinical trials, until we are able to obtain sufficient financing. If such sufficient financing is not received timely, we would then need to pursue a plan to license or sell our assets, seek to be acquired by another entity, cease operations and/or seek bankruptcy protection.
Cash Flows
The following table summarizes our consolidated statement of cash flows for the years ended December 31, 2016, 2017 and 2018.
| Year ended December 31, | ||||||||||||||||
| 2016 | 2017 | 2018 | 2018 | |||||||||||||
| (NIS in thousands) | (Convenience translation into USD in thousands(1)) |
|||||||||||||||
| Net cash provided by (used in): | ||||||||||||||||
| Operating activities | (19,357 | ) | (17,884 | ) | (4,665 | ) | (1,245 | ) | ||||||||
| Investing activities | (492 | ) | (447 | ) | (3,603 | ) | (962 | ) | ||||||||
| Financing activities | 18,486 | 32,395 | 9,915 | 2,647 | ||||||||||||
| (1) | Calculated using the exchange rate reported by the Bank of Israel for December 31, 2018, at the rate of one U.S. dollar per NIS 3.748. |
88
Net Cash Used in Operating Activities
The use of cash in all periods resulted primarily from our net losses adjusted for non-cash charges and measurements and changes in components of working capital. Adjustments to net income for non-cash items include depreciation and amortization and share-based compensation.
Net cash used in operating activities resulted primarily from our net losses adjusted for non-cash charges and measurements and changes in components of working capital. Adjustments to net loss for non-cash items include depreciation and amortization, equity-based compensation and exchange differences on cash and cash equivalents. This cash flow mainly reflects the cash needed for funding the products and pipeline products development and management costs of the Company during the applicable periods.
Net cash used in operating activities in the year ended December 31, 2018 totaled NIS 4.7 million ($1.2 million) and consisted primarily of (i) a net loss of NIS 13.9 million ($3.7 million), adjusted for non-cash items including depreciation and amortization of NIS 1.5 million ($392,000) and shared-based compensation of NIS 5.2 million, ($1.4 million), and (ii) a net increase in operating assets and liabilities of NIS 4.1 million ($1.1 million), which are mainly attributable to an increase in contract liabilities of NIS 7.3 million ($1.9 million) relating to the United License Agreement, and an increase in inventory of NIS 2.4 million ($628,000) and an increase in trade receivables of NIS 1.6 million ($421,000), all as an outcome of the growth in production and sales activity of BioInk.
Net cash used in operating activities in the year ended December 31, 2017 totaled NIS 17.9 million ($5.2 million) and consisted primarily of (i) a net loss of NIS 20.9 million ($6.0 million), adjusted for non-cash items, including depreciation and amortization of NIS 1.0 million ($288,000) and share based compensation of NIS 5.0 million ($1.4 million), and (ii) a net increase in operating assets and liabilities of NIS 3.1 million ($894,000), which are mainly attributable to a decrease in royalties to the IIA liability of NIS 1.0 million ($288,000), and a decrease in trade payables and long term payable of NIS 2.2 million ($635,000) as a result of the decrease in our development activity with the Surgical Matrix Carrier.
Net cash used in operating activities in the year ended December 31, 2016 totaled NIS 19.3 million ($5.0 million) and consisted primarily of a net loss of NIS 27.9 million ($7.3 million), adjusted for non-cash items including depreciation and amortization of NIS 864,000 ($225,000) and share based compensation of NIS 3.6 million ($936,000), and a net decrease in operating assets and liabilities of NIS 3.9 million ($1.0 million), mainly attributable to an increase in royalties to the IIA liability of NIS 2.2 million ($572,000), an increase in trade payables and long term payable of NIS 2.5 million ($650,000), all as a result of an increase of our development activity with VergenixSTR and the Surgical Matrix Carrier and an increase in inventory of NIS 487,000 ($127,000 million).
Net Cash Used in Investing Activities
Net cash used in investing activities was NIS 3.6 million ($1.0 million) during the year ended December 31, 2018 and NIS 447,000 ($129,000) during the year ended December 31, 2017. The increase in the amount of approximately NIS 3.2 million ($854,000) relates mainly to the purchases of property and equipment in the amount of NIS 3.0 million ($796,000) for the establishment of our production facility in Rehovot, and restricted deposit in the amount of NIS 620,000 ($166,000) against the lease of our new office and labs facility in Rehovot Israel.
Net cash used in investing activities was NIS 447,000 ($129,000) and NIS 492,000 ($128,000) for the year ended December 31, 2017 and 2016, respectively, and related primarily to the purchases of property and equipment.
Net Cash Provided by Financing Activities
Net cash provided by financing activities amounted to approximately NIS 9.9 million ($2.6 million) for 2018 and NIS 32.4 million ($9.3 million) in 2017. In 2018, we consummated equity raises of net NIS 10.0 million ($2.7 million) in return for proceeds from the issuances of securities under the Alpha Purchase Agreement and several private investments of our existing shareholders. In addition we received, net of returns, NIS 168,000 ($46,000) from a loan and made payments of NIS 252,000 ($67,000) for equipment on financing terms.
Cash flow from financing activities in 2017 amounted to NIS 32.4 million ($9.3 million) mainly in return for the issuance of our securities under private purchase agreements, public equity raise and the exercise of warrants.
89
Net cash provided by financing activities was NIS 32.4 million ($9.3 million) for the year ended December 31, 2017, compared to NIS 18.5 million ($4.8 million) in the year ended December 31, 2016. Proceeds generated by 2017 financing includes NIS 6.8 million ($2.0 million) in return for the issuance of our ordinary shares and warrants in an equity raise in Israel, NIS 3.6 million ($1.0 million) for exercise of warrants, and NIS 22.2 million ($6.4 million) of proceeds from the issuances of securities under the Alpha Purchase Agreement, the Meitav Purchase Agreement, and the Sagy Purchase Agreement, and net of NIS 253,000 ($73,000) of payment made for equipment on financing terms. Cash flow from financing activities in the year ended December 31, 2016 amounted to NIS 18.5 million ($4.8 million) in return for the issuance of our ordinary shares and warrants in an equity raise in Israel.
Cash and Funding Sources
The table below summarizes our sources of funding for the years ended December 31, 2016, 2017 and 2018:
| Issuance of Ordinary Shares and Warrants |
Government Grants and Strategic Collaboration |
Total | Total | |||||||||||||
| (NIS in thousands) | (Convenience translation into USD in thousands(1)) |
|||||||||||||||
| Year ended December 31, 2018 | 9,999 | 321 | 10,320 | 2,753 | ||||||||||||
| Year ended December 31, 2017 | 32,648 | 2,044 | 34,692 | 9,256 | ||||||||||||
| Year ended December 31, 2016 | 19,702 | 12,411 | 32,113 | 8,568 | ||||||||||||
| (1) | Calculated using the exchange rate reported by the Bank of Israel for December 31, 2018 at the rate of one U.S. dollar per NIS 3.748. |
Funding Requirements
We believe that our existing cash and cash equivalents will enable us to fund our operating expenses and capital expenditures into the first quarter of 2020. We have based this estimate on assumptions that may prove to be wrong, and we could use our capital resources sooner than we currently expect.
Our present and future funding requirements will depend on many factors, including, among other things:
| ● | the progress, timing, and completion of preclinical testing and clinical trials in the U.S. for tissues and organs which are based on our BioInk, VergenixSTR and VergenixFG or any future pipeline product; |
| ● | selling and marketing activities undertaken in connection with the commercialization of VergenixSTR and VergenixFG and any other products; |
| ● | the costs of upscaling our manufacturing capabilities; |
| ● | costs involved in the development of distribution channels, and for an effective sales and marketing organization, for the commercialization of our products in Europe; |
| ● | the time and costs involved in obtaining regulatory approvals and any delays we may encounter as a result of evolving regulatory requirements or adverse results with respect to any of these products; |
| ● | the number of potential new products we identify and decide to develop; and |
| ● | the costs involved in filing patent applications and maintaining and enforcing patents or defending against claims or infringements raised by third parties. |
90
For more information as to the risks associated with our future funding needs, see “Item 3.D. Risk Factors—We will need to raise additional funding, which may not be available on acceptable terms, or at all. Failure to obtain additional capital when needed may force us to delay, limit, or terminate our product development efforts or other operations.”
| C. | Research and Development, Patents and Licenses |
See above, under Item 5A – “Operating Results.”
| D. | Trend Information |
We are in a development stage with regard to different 3D BioInks and are in early stages of commercialization for VergenixFG and VergenixSTR in Europe, and our BioInks for customers that develop technologies for 3D bio-printing of tissues and organs. It is not possible for us to predict with any degree of accuracy the outcome of our research, development, or commercialization efforts. As such, it is not possible for us to predict with any degree of accuracy any known trends, uncertainties, demands, commitments or events that are reasonably likely to have a material effect on our net sales or revenues, income from continuing operations, profitability, liquidity or capital resources, or that would cause reported financial information to not necessarily be indicative of future operating results or financial condition. However, to the extent possible, certain trends, uncertainties, demands, commitments and events are in this “Operating and Financial Review and Prospects.”
| E. | Off-Balance Sheet Arrangements |
As of December 31, 2018, we do not have any, and during the periods presented, we did not have any, off-balance sheet arrangements.
| F. | Contractual Obligations |
Our significant contractual obligations as of December 31, 2018 are summarized in the following table.
| Payments due by period | ||||||||||||||||||||
| Less than 1 year |
1 to 2 years | 2 to 5 years | More than 5 years |
Total | ||||||||||||||||
| (NIS in thousands) | ||||||||||||||||||||
| Operating lease obligations(1) | 2,011 | 2,166 | 5,749 | 8,070 | 17,996 | |||||||||||||||
| (1) | Operating lease obligations consist of payments pursuant to lease agreements for office and laboratory facilities, as well as lease agreements for six vehicles, which generally run for a period of three years. |
Our balance sheet liabilities do not include all of the obligations regarding royalties that we are obligated to pay to the IIA based on future sales of our products. As of December 31, 2018, our balance sheet liability in amount of NIS 1.2 million includes the liability for future royalties payable to the IIA where the maximum royalty amount that would be payable by us, before interest, is approximately NIS 31.8 million (assuming 100% of the royalties are payable). This liability is contingent upon sales of our rhCollagen-based products.
91
| ITEM 6. | DIRECTORS, SENIOR MANAGEMENT AND EMPLOYEES |
| A. | Directors and Senior Management |
The following table sets forth certain information relating to our directors and senior management. Unless otherwise stated, the address for our directors and senior management is at the Company’s registered address c/o 4 Oppenheimer, Weizmann Science Park, P.O. Box 4132, Rehovot 7670104, Israel.
| Name | Age | Position | ||
| Senior Management | ||||
| Yehiel Tal | 66 | Chief Executive Officer | ||
| Prof. Oded Shoseyov (1) | 62 | Founder, Chief Scientist | ||
| Eran Rotem, CPA | 51 | Deputy CEO and Chief Financial Officer | ||
| Dr. Ilana Belzer | 59 | Chief Operating Officer | ||
| Dr. Nadav Orr | 61 | Vice President, Research and Development | ||
| Dr. Philippe Bensimon | 53 | Vice President, Regulatory Affairs and Quality Assurance | ||
| Non-Employee Director | ||||
| Jonathan M.N. Rigby(6)(8) | 51 | Chairman of the Board and Director | ||
| Adi Goldin | 44 | Director | ||
| Dr. Abraham Havron(2)(5)(6)(7)(8) | 71 | Director | ||
| Dr. Gili Hart(2)(3)(4)(5)(6)(7)(8) | 44 | Director | ||
| Scott R. Burell(3)(4)(5)(7)(8) | 54 | Director | ||
| Dr. Elan Penn(2)(3)(4)(5)(6)(7)(8) | 67 | Director | ||
| Dr. Wolfgang Ruttenstorfer(6)(8) | 68 | Director |
| (1) | On March 26, 2019, Prof. Shoseyov became our Chief Scientist instead of our Chief Scientific Officer and is no longer considered an office holder under the Companies Law. |
| (2) | Member of the Compensation Committee |
| (3) | Member of the Audit Committee |
| (4) | Member of Financial Statements Committee |
| (5) | External Director under Israeli Law |
| (6) | Independent Director under Israeli Law |
| (7) | Member of the Nominating and Corporate Governance Committee |
| (8) | Independent Director under the Nasdaq Listing Rules |
Senior Management
Yehiel Tal has served as our chief executive officer since January 2010. Mr. Tal possesses over 30 years of management experience in the Israeli and American high-tech and biotechnology industries. Prior to joining us, Mr. Tal was the chief executive officer and co-founder of Regentis Biomaterials Ltd. Prior to that Mr. Tal served as vice-president of business development at ProChon BioTech Ltd. He has also served as vice president of marketing and business development at OrthoScan Technologies Ltd. and director of business development and business unit manager at Kulicke and Soffa Industries, Inc. Mr. Tal holds a Bachelor’s and a Master’s degree in mechanical engineering from the Technion, Israel Institute of Technology.
Prof. Oded Shoseyov founded our subsidiary CollPlant Ltd. in 2004, and currently serves as our Chief Scientist since March 2019. Prof. Shoseyov served as our Chief Scientific Officer from August 2008 until March 2019, and a member of our board of directors from May 2010 until October 2016. Prof. Shoseyov is a faculty member of the Hebrew University of Jerusalem. He has extensive experience with plant transformation systems and protein engineering. Prof. Shoseyov has authored or co-authored over 160 scientific publications and is the inventor or co-inventor of 45 patents. Prof. Shoseyov holds a Ph.D. from The Hebrew University of Jerusalem, Israel. Prof. Shoseyov received the Outstanding Scientist Polak Award for 2002, the 1999 and 2010 Kay Awards for Innovative and Applied Research, and The 2012 Israel Prime Minister Citation for Entrepreneurship and Innovation. He is the scientific founder of nine companies, including: SP-Nano Ltd., a nano-biotech company which manufactures SP1-Carbon Nano Tube coated fabrics for the composite industry; CBD-Technologies/FuturaGene, a forestry agro-biotech company that develops and commercializes transgenic trees for the pulp and paper and the bio-fuel industry; Melodea Ltd., a nano-biotech company that develops and manufactures Nano Crystaline Cellulose from sludge for structural foam additives for the paint, printing and packaging industries; and Valentis Nanotech Ltd., a nanotechnology company that develops and manufactures nano-bio-based transparent films for food packaging and agriculture.
92
Eran Rotem has served as our chief financial officer since January 2012 and, since November 2017, also as our deputy CEO. Mr. Rotem possesses more than 23 years of broad financial and operational experience, primarily with biotechnology and industrial companies. Prior to joining us, Mr. Rotem served as the chief financial officer of Tefron Ltd., an industrial global company traded on both the Tel Aviv Stock Exchange (TASE:TFRN) and on the OTCBB (OTC:TFRFF) in the United States. Before Tefron, Mr. Rotem served as chief financial officer of Healthcare Technologies, Ltd. (NASDAQ:HCTL) and Gamida Ltd., a group of companies that specialize in the development, manufacturing, and marketing of clinical diagnostic test kits, as well as medical equipment and services to the biotechnology and high-tech industries. Prior to joining Healthcare Technologies, Ltd., Mr. Rotem served as a senior manager at Ernst & Young. Mr. Rotem holds a Bachelor’s degree in Accounting and Business Administration from the Tel Aviv College of Management and is a Certified Public Accountant in Israel.
Dr. Ilana Belzer has served as our chief operating officer since October 2015. Prior to joining us, Dr. Belzer served as the chief operating officer of BioHarvest, an innovative biotechnology company, from October 2012 to September 2015, and prior to that as vice president of research and development and operations at Procognia Ltd. Prior to that, Dr. Belzer held executive positions in Omrix Biopharmaceuticals Inc., now part of the Johnson & Johnson family of companies, and InterPharm Laboratories Ltd., now a subsidiary of Merck-Serono. Dr. Belzer holds an M.Sc., a B.Sc. and a Ph.D. in Microbiology and Cell Biology from Tel Aviv University, Israel.
Dr. Nadav Orr has served as our vice president of research and development since September 2014. Dr. Orr has over 16 years of experience in research and development, including 12 years in the development of biosurgery products. Prior joining us, Dr. Orr served as the associate director of research and development at Omrix Biopharmaceuticals Ltd., a subsidiary of Ethicon US LLC, part of the Johnson & Johnson family of companies. As part of his role at Omrix, Dr. Orr led an international team in the development of hemostatic combination products and led base business support for production processes and products. Dr. Orr holds a Ph.D. from the Weizmann Institute of Science, Israel.
Dr. Philippe Bensimon has served as our vice president of quality assurance and clinical affairs since February 2011. Dr. Bensimon has 26 years of experience in regulatory affairs, quality assurance and clinical affairs in international medical device companies. Prior to joining us Dr. Bensimon served for 14 years at InterVascular Datascope (now Maquet-Getinge Group), a manufacturer of long-term cardiovascular implants, as director of regulatory affairs, quality assurance, and clinical affairs. Dr. Bensimon also served for five years at 3M Medical as manager of regulatory affairs. Dr. Bensimon holds a PharmD degree from the University of Pharmacy, Marseille, France.
Non-Employee Directors
Jonathan M.N. Rigby has served on our board of directors as chairman since February 2019. Mr. Rigby has served as the President, CEO and a member of the Board of Directors of SteadyMed Ltd, a specialty pharmaceutical company focused on Pulmonary Arterial Hypertension, since August 2011. In 2015, Mr. Rigby led the listing of SteadyMed on Nasdaq; STDY. In August 2018, Mr. Rigby led and closed the sale of SteadyMed to United Therapeutics Corporation (Nasdaq; UTHR). Mr. Rigby currently serves as the President & CEO of SteadyMed, a United Therapeutics Corporation. Mr. Rigby joined the Board of Directors of Xeris Pharmaceuticals in March 2016 and was instrumental in the growth of the company leading to its IPO on Nasdaq (XERS) in June 2018. Mr. Rigby currently serves on the Board of Directors of Xeris Pharmaceuticals and is a member of the Audit Committee and the Chair of the Nominating and Governance Committee. In 2006, Mr. Rigby cofounded Zogenix, Inc., a specialty pharmaceutical company focused on the development and commercialization of CNS and pain products, where he served as the company’s Vice President of Business Development until December 2010. As a member of the senior management team he played an important role in the Nasdaq listing of Zogenix (Nasdaq; ZGNX) in 2010. Between 2002 and 2006, Mr. Rigby held positions of increasing responsibility at Aradigm Corporation, including Vice President of Business Development where he was involved in M&A activities as well as inhalation delivery technology licensing in various therapeutic fields including Pulmonary Arterial Hypertension, or PAH. Between 1995 and 2002, Mr. Rigby held various commercial and business development positions at Profile Therapeutics, UK, where he played a key role in the licensing of inhalation technology that resulted in the approval and launch of an inhalation product to treat PAH. Between 1990 and 1995, he held various sales and marketing positions at large pharmaceutical companies including Merck Sharpe and Dohme, or MSD, and Bristol Myers Squibb, or BMS. Mr. Rigby has a Bachelor of Science Degree, with Honors, in Biological Sciences from Sheffield University, UK, and an MBA from Portsmouth University, UK.
93
Adi Goldin has served on our board of directors since May 2010, and from May 2016 to January 2018, acted as our chairman. Mr. Goldin has over 15 years of experience in the life science, industrial, and technology industries in the areas of investments, business strategy, deal structure, and company management. For the last 13 years, Mr. Goldin has served as a vice president at Docor International BV and has played a key role in investing, managing, and nurturing technology-driven companies and startups in the information technology, industrial, and life science industries. Mr. Goldin also serves on the board of several portfolio companies of Docor. Until 2010, Mr. Goldin was the chief executive officer of Softlib Ltd., an information technology company. Previously, Mr. Goldin was VP of investments and analysis at Inventech Investment Company Ltd. (TASE: IVTC), where he took an active role in building startup companies and was involved in public offerings, M&A, and various aspects of the capital markets. In addition, Mr. Goldin was part of the teaching staff of the Executive MBA program run by Tel Aviv University. Mr. Goldin participated in the International Marketing and Global Consulting Program, a joint project of the University of Pennsylvania’s Wharton Business School and Tel Aviv University’s Business School. Mr. Goldin is a member of the Israel Bar Association. Mr. Goldin holds Bachelor’s and Master’s degrees in economics, summa cum laude, and an LL.B. in law from Tel Aviv University, Israel.
Dr. Abraham Havron has served on our board of directors since May 2016. Dr. Havron is a 38-year veteran of the biotechnology industry. Between 2011 and 2018, Dr. Havron served as a director at Kamada (NASDAQ: KMDA) where he was initially elected as an external director (within the meaning of the Companies Law) and served in such capacity until January 30, 2017, since which time he has served as an ordinary (non-external) director. From 2005 to 2013, Dr. Havron has served as the Chief Executive Officer and a director of PROLOR Biotech Ltd., which in 2013 merged with OPKO Health Inc. Dr. Havron was a member of the founding team and Director of Research and Development of Interpharm Laboratories Ltd. (a subsidiary of Merck Serono S.A.) from 1980 to 1987. Dr. Havron served as Vice-President of Manufacturing and Process-Development of BioTechnology General Ltd., based in Rehovot, Israel (now, a subsidiary of Ferring Pharmaceuticals) from 1987 to 1999, and Vice President and Chief Technology Officer of Clal Biotechnology Industries Ltd. from 1999 to 2003. Since 2014, Dr. Havron has also served on the board of directors of MediWound Ltd. (NASDAQ: MDWD) until June 2017 and Enlivex Theraputics Ltd., a private company. Dr. Havron earned his PhD in Bio-Organic Chemistry from the Weizmann Institute of Science and served as a Research Fellow at the Harvard Medical School, Department of Radiology.
Dr. Gili Hart has served on our board of directors since July 2017. Dr. Hart served as the Chief Executive Officer of OPKO Biologics from 2014 and until 2017. From 2011 to 2014, Dr. Hart served as Vice President of Prolor Biotech Ltd. Dr. Hart serves as a director in Enlivex Therapeutics, and Dr. Hart holds a B.Sc degree in Biological engineering and an M.Sc degree from the Weizmann Institute of Science as well as a Ph.D. from the Weizmann Institute of Science.
Scott R. Burell has served on our board of directors since October 2017. Since August 2018, Mr. Burell serves as the Chief Financial Officer of AIVITA Biomedical Systems, Inc., a private biopharmaceutical company. From November 2006 until November 2017, Mr. Burell served as Chief Financial Officer, Secretary and Treasurer of CombiMatrix Corporation (NASDAQ: CBMX). Prior to this, Mr. Burell had served as CombiMatrix’s Vice President of Finance since November 2001 and as its Controller from February 2001 to November 2001. From May 1999 to first joining CombiMatrix in February 2001, Mr. Burell was the Controller for Network Commerce, Inc., a publicly traded technology and information infrastructure company located in Seattle. Prior to this, Mr. Burell spent nine years with Arthur Andersen’s Audit and Business Advisory practice in Seattle. During his tenure in public accounting, Mr. Burell worked with many clients, both public and private, in the high-tech and healthcare markets, and was involved in numerous public offerings, spin-offs, mergers and acquisitions. Mr. Burell is also a member of the Board of Directors of Microbot Medical (NASDAQ: MBOT), an Israeli-based medical device company specializing in the researching, designing, developing and commercializing of transformational micro-robotics medical technologies, and of MER Telemanagement Solutions Ltd., an Israeli-based telecommunications services company. Mr. Burell obtained his Washington state CPA license in 1992 and is a certified public accountant (currently inactive). He holds Bachelor of Science degrees in Accounting and Business Finance from Central Washington University.
94
Dr. Elan Penn has served on our board of directors since January 2018. Dr. Penn has served as chief executive officer and chairman of Penn Publishing Ltd., a private company based in Tel Aviv, Israel, since 2001. From 2000 to 2001, Dr. Penn served as vice president of finance and administration of A.I. Research and Development Ltd. Dr. Penn served as chief executive officer of Sivan Computer Training Company Ltd. during the years 1998 through 2000. From 1992 to 2000, Dr. Penn served as vice president of finance and administration of Mashov Computers Ltd. From 1987 to 1991 and again from 1992 to 1997, Dr. Penn served as vice president of finance and administration of Magic Software Enterprises Ltd. (NASDAQ: MGIC) and, from 2005 to 2014, served as an external director of Magic Software. Dr. Penn previously served as a director of Telkoor Power Supplies Ltd. (TASE: TLCR) and Nexgen Biofuels Ltd. (formerly Healthcare Technologies Ltd) (OTC: NXGN). Dr. Penn holds a B.A. degree in Economics from the Hebrew University of Jerusalem and a Ph.D. in Management Science from the University of London.
Dr. Wolfgang Ruttenstorfer has served on the Board of Directors since July 2018. Beginning in 1976, Dr. Ruttenstorfer spent a total of approximately 30 years in various capacities with OMV AG, an integrated oil and gas company headquartered in Vienna, Austria, culminating in his position as chairman of the executive board from 2002 through mid-2011. From 1997 through 1999, Dr. Ruttenstorfer stepped away from OMV to serve as Deputy Finance Minister of Austria. Since 2012, Dr. Ruttenstorfer has been a board member of NIS a.d. Novi Sad, one of the largest, vertically integrated energy companies in southeast Europe, as well as a member of the supervisory board of RHI AG, a leading global supplier of high-grade refractory products, systems and services. Since 2011, he has also served as a member of the supervisory board of Flughafen Wien, AG, the Vienna International Airport. From 2007 through 2011, Dr. Ruttenstorfer lent his expertise to Roche Holding AG, as a member of the board of directors. During the period from 2009 to early 2018, Dr. Ruttenstorfer also served at various times on a number of other supervisory boards, including that of CA Immobilien AG, Telekom Austria AG, and Vienna Insurance Group AG. Dr. Ruttenstorfer received his doctorate from the University of Vienna in Economics and Business.
Adi Goldin and Dr. Havron are also board members of CollPlant Ltd., our wholly owned subsidiary.
Advisory Boards
We have established a scientific advisory board and a clinical advisory board. The members of our advisory boards are appointed by our chief executive officer after consultation with our board of directors. Once nominated, the members of our advisory boards sign a standard letter of engagement. Most of the members of our advisory boards are not appointed for a specific term and their position may be terminated by either us or the member of the advisory board according to standard notice periods. The members of our advisory boards are all paid either daily or hourly fees for their services and are entitled to the reimbursement of their expenses. Furthermore, several of the members of our advisory boards have been granted options due to their strategic role and years of service. The members of our advisory boards are as follows:
Scientific Advisory Board
Prof. Avraham Hershko
Prof. Shay Soker
Prof. Vicki Rosen
Prof. Abhay Pandit
Prof. Ofer Levy, MD, MCh (Orth)
Joseph M. Lane, MD
Clinical Advisory Board
Joseph M. Lane, MD
Scott Rodeo, MD
Thomas Serena, MD
Gabi Agar, MD
Prof. Ofer Levy, MD, MCh (Orth)
95
| B. | Compensation |
Compensation of Senior Management and Directors
The following table presents in the aggregate all compensation we paid to all of our senior management and directors as a group for the year ended December 31, 2018. The table does not include any amounts we paid to reimburse any of such persons for costs incurred in providing us with services during this period.
| Salaries, fees, commissions, and bonuses(1)(2) (thousand NIS) |
Salaries, fees, commissions, and bonuses(1)(2) (4) (thousand USD) |
Value of Options Granted(3) (thousand NIS) |
Value of Options Granted(3)(4) (thousand USD) |
|||||||||||||
| All senior management and directors as a group, consisting of 13 persons | 6,269 | 1,673 | 2,680 | 715 | ||||||||||||
| (1) | Salary includes cost of salary to the Company and ancillary benefits such as payments to the National Insurance Institute, advanced education funds, managers’ insurance and pension funds; vacation pay; recuperations pay as mandated by Israeli law. |
| (2) | Includes cost of salary to two former executive officers who ended their tenure during 2018. |
| (3) | Consists of amounts recognized as share-based compensation expense for the year ended December 31, 2018. Assumptions and key variables used in the calculation of such amounts are discussed in Note 14 of our financial statements. |
| (4) | Calculated using the exchange rate reported by the Bank of Israel for December 31, 2018, at the rate of one U.S. dollar per NIS 3.748. |
In accordance with the Companies Law, the following table presents information regarding compensation of our five most highly paid office holders, namely our Chief Executive Officer, deputy CEO and Chief Financial Officer, Vice President Regulatory Affairs and Quality Assurance, Vice President Research and Development and during the year ended December 31, 2018.
| Name and Position | Salary(1) (thousand NIS) |
Bonus (thousand NIS) |
Consulting Fees (thousand NIS) |
Value of Options Granted(2) (thousand NIS) |
Total (thousand NIS) |
Total (thousand US dollar)(3) |
||||||||||||||||||
| Yehiel Tal, CEO | 1,076 | 260 | - | 793 | 2,129 | 568 | ||||||||||||||||||
| Eran Rotem, Deputy CEO & CFO | 951 | 216 | - | 345 | 1,512 | 403 | ||||||||||||||||||
| Oded Shoseyov (4) | - | - | 384 | 562 | 946 | 252 | ||||||||||||||||||
| Philippe Bensimon, VP Reg. Affairs & QA | 665 | 44 | - | 120 | 829 | 221 | ||||||||||||||||||
| Nadav Orr, VP R&D | 700 | 80 | - | 116 | 896 | 239 | ||||||||||||||||||
| (1) | Salary includes cost of salary to the Company and ancillary benefits such as payments to the National Insurance Institute, advanced education funds, managers’ insurance and pension funds; vacation pay; recuperations pay as mandated by Israeli law. |
| (2) | Consists of amounts recognized as share-based compensation expense for the year ended December 31, 2018. Assumptions and key variables used in the calculation of such amounts are discussed in Note 14 of our financial statements. |
| (3) | Calculated using the exchange rate reported by the Bank of Israel for December 31, 2018, at the rate of one U.S. dollar per NIS 3.748. |
| (4) | On March 26, 2019, Prof. Shoseyov became our Chief Scientist instead of our Chief Scientific Officer and is no longer considered an office holder under the Companies Law. |
96
Compensation of Directors
Under the Companies Law and the rules and regulations promulgated thereunder, external directors are generally entitled to fixed annual compensation and an additional payment for each meeting attended. We currently pay our directors an annual fee of NIS 29,000 and a per meeting fee of NIS 1,800.
During 2018, we granted Dr. Elan Penn, Dr. Gili Hart, Dr. Abraham Havron and Scott Burell 500,000 options to purchase 500,000 ordinary shares each and, to Adi Goldin 650,000 options to purchase 650,000 ordinary shares, each at an exercise price per option of NIS 0.58 ($0.15). The options vest subject to a vesting period of four years, with a quarter of the options vesting on the first anniversary of the grant date, and the remaining options vesting in equal parts at the end of every quarter thereafter
In January 2019, we granted, subject to shareholders approval, Dr. Elan Penn, Dr. Gili Hart, Dr. Abraham Havron, Scott Burell and Adi Goldin 250,000 options to purchase 250,000 ordinary shares each and, to Dr. Wolfgang Ruttenstorfer 750,000 options to purchase 750,000 ordinary shares each, and to Jonathan M.N. Rigby 12,319,500 options to purchase 12,319,500 ordinary shares, each at an exercise price per option of $0.101. The options vest subject to a vesting period of four years, with a quarter of the options vesting on the first anniversary of the grant date, and the remaining options vesting in equal parts at the end of every quarter thereafter.
Employment and Services Agreements with Senior Management
Yehiel Tal
Mr. Tal has been our chief executive officer since January 2010. Mr. Tal’s current gross monthly salary is NIS 65,000. In addition, Mr. Tal is entitled to social benefits, such as paid annual vacation days, severance pay, annual recreation allowance, manager’s insurance, sick leave, education fund and expenses reimbursement. In addition, we provide Mr. Tal with company’s car and a mobile phone. Mr. Tal’s employment agreement is terminable by either us or Mr. Tal upon 90 days’ prior written notice other than in the case of a termination for cause. Mr. Tal’s employment agreement contains a non-compete obligation for a period of 12 months following termination of his employment, customary provisions regarding confidentiality of information and assignment of inventions. The agreement does not provide for benefits upon the termination of employment, other than payment of salary and benefits during the required notice period. Mr. Tal’s agreement also provides for annual bonus payments based upon criteria determined by the board of directors, as well as special bonuses which may be payable upon the achievement of specified milestones, such as the execution of an income-generating commercial agreement or consummation of an initial public offering (subject to the satisfaction of certain conditions). Mr. Tal’s bonus for the year 2018 was based on Mr. Tal’s achievement of objectives which includes business development and positioning of 3D BioInk as the Company’s growth engine, and for his significant contribution to the Company’s engagement with United Therapeutics Corporation in United License Agreement for 3D bio-printing of solid-organ scaffolds for human transplants. As of March 15, 2019, Mr. Tal held 1,505,875 ordinary shares and 9,573,041 options to purchase 5,691,014 ordinary shares at a weighted average exercise price of NIS 0.99, of which 6,406,166 options are fully vested and 354,375 options will vest by May 19, 2019, and 2,812,500 options will vest over a period of three years from January 14, 2019, in equal parts at the end of every quarter thereafter. In addition, the board of directors approved a grant to Mr. Tal of 2,700,000 options to purchase 2,700,000 ordinary shares, subject to shareholders approval, which will vest over a period of four years with a quarter of the options vesting on January 30, 2020, and the remaining options vesting in equal parts at the end of every quarter thereafter.
97
Eran Rotem
Mr. Rotem has served as our chief financial officer since January 2012 and since November 2017, also as our deputy chief executive officer. Mr. Rotem’s current gross monthly salary is NIS 54,000. In addition, Mr. Rotem is entitled to social benefits, such as paid annual vacation days, severance pay, annual recreation allowance, manager’s insurance, sick leave, education fund and expenses reimbursement. In addition, we provide Mr. Rotem with a leased company car and a mobile phone. Mr. Rotem’s employment agreement is terminable by either us or Mr. Rotem upon 90 days’ prior written notice. Mr. Rotem’s employment agreement contains a non-compete obligation for a period of 12 months following termination of his employment and customary provisions regarding confidentiality of information and assignment of inventions. Mr. Rotem’s employment agreement also provides for a grant of options to purchase up to 150,000 ordinary shares under the 2010 Plan, which will vest subject to certain conditions. The agreement does not provide for benefits upon the termination of employment, other than payment of salary and benefits during the required notice period. Mr. Rotem’s agreement also provides for annual bonus equal to up to two months’ salary based upon successful achievement of objectives determined by our chief executive officer and, in accordance with our compensation policy, within three months from the beginning of each calendar year and approval of our board of directors. Mr. Rotem’s bonus for the year 2018 was based on Mr. Rotem’s achievement of objectives, which includes planning and execution of 2018 fundraising rounds and his role in the United License Agreement. As of March 15, 2019, Mr. Rotem held 7,826,607 options to purchase 5,442,202 ordinary shares at a weighted average exercise price of NIS 0.77, of which 3,951,607 options are fully vested, and 187,500 options will vest by May 19, 2019, and 1,687,500 options will vest over a period of three years from December 27, 2018, in equal parts at the end of every quarter thereafter and 2,000,000 options will vest over a period of four years, with a quarter of the options vesting on January 30, 2020, and the remaining options vesting in equal parts at the end of every quarter thereafter.
Prof. Oded Shoseyov
Prof. Shoseyov founded our subsidiary CollPlant Ltd. in 2004, and currently serves as our Chief Scientist since March 2019. Prof. Shoseyov served as our Chief Scientific Officer between August 2008 and March 2019. We entered into written consulting and option agreements with Prof. Shoseyov, and he is currently paid a monthly service fee of NIS 32,000, including VAT. Prof. Shoseyov’s consulting agreement creates an independent contractor relationship between us and therefore does not provide for severance or other employment related benefits. Prof. Shoseyov’s agreement is terminable by either us or Prof. Shoseyov upon 90 days’ prior written notice other than in the case of a termination for cause. Under the provisions of the services agreement we have complete ownership in any invention which is derived from our operations and businesses as well as first rights (for the development and commercialization) in any invention that is not our invention and that may be a result of Prof. Shoseyov’s activity in the course of providing the services with the exceptions of specific inventions defined in the agreement. The services agreement sets a non-compete obligation for a period of two years following the later of the termination of the services agreement, disposal of all of our securities held by Prof. Shoseyov, the termination of Prof. Shoseyov’s membership in our board of directors or termination of any other of Prof. Shoseyov’s engagement with us, and further provisions regarding confidentiality. As of March 15, 2019, Prof. Shoseyov held 2,737,573 ordinary shares and 14,367,904 options to purchase 6,122,635 ordinary shares at a weighted average exercise price of NIS 1.4, of which 10,117,904 options are fully vested and 2,500,000 options will vest over a period of five years from September 22, 2016, in equal parts at the end of every quarter thereafter and 750,000 options will vest over a period of three years from December 27, 2018 in equal parts at the end of every quarter thereafter and 1,000,000 options will vest over a period of four years, with a quarter of the options vesting on January 30, 2020, and the remaining options vesting in equal parts at the end of every quarter thereafter.
Dr. Philippe Bensimon
Dr. Bensimon has served as our vice president of regulatory affairs quality assurance and clinical affairs since February 2011. Dr. Bensimon is entitled to a monthly gross salary of NIS 44,000, and to social benefits, such as paid annual vacation days, severance pay, annual recreation allowance, manager’s insurance, sick leave, education fund and expenses reimbursement. In addition, we provide Dr. Bensimon with a leased company car and a mobile phone. Dr. Bensimon’s employment agreement is terminable by either us or Dr. Bensimon upon 60 days’ prior written notice other than in the case of a termination for cause. Dr. Bensimon’s employment agreement contains a non-compete obligation for a period of 12 months following termination of his employment and customary provisions regarding confidentiality of information and assignment of inventions. Dr. Bensimon’s employment agreement also provides for a grant of options to purchase up to 66,667 ordinary shares under the 2010 Plan, which will vest subject to certain conditions. The agreement does not provide for benefits upon the termination of employment, other than payment of salary and benefits during the required notice period. Dr. Bensimon’s agreement also provides for annual bonus payments based upon successful achievement of objectives determined each year by our chief executive officer and in accordance with our compensation policy and approval of our board of directors. Dr. Bensimon’s bonus for the year 2018 was based on Dr. Bensimon’s achievement of objectives which includes product regulation process, and planning and execution of the post marketing surveillance studies in Europe, with regards to the Company’s products. As of March 15, 2019, Dr. Bensimon held 3,400,000 options to purchase 2,166,667 ordinary shares at a weighted exercise price of NIS 0.9, of which 1,953,125 options are fully vested, and 84,375 options will vest by May 19, 2019, and 562,500 options will vest over a period of three years from December 27, 2018, in equal parts at the end of every quarter thereafter and 800,000 options will vest over a period of four years, with a quarter of the options vesting on January 30, 2020, and the remaining options vesting in equal parts at the end of every quarter thereafter.
98
Dr. Ilana Belzer
Dr. Belzer has served as our chief operating officer since October 2015. Dr. Belzer is entitled to a monthly gross salary of NIS 43,000, and to social benefits, such as paid annual vacation days, severance pay, annual recreation allowance, manager’s insurance, sick leave, education fund and expenses reimbursement. In addition, we provide Dr. Belzer with a leased company car and a mobile phone. Dr. Belzer’s employment agreement is terminable by either us or Dr. Belzer upon 60 days’ prior written notice. Dr. Belzer’s employment agreement contains a non-compete obligation for a period of six months following termination of her employment and customary provisions regarding confidentiality of information and assignment of inventions. The agreement does not provide for benefits upon the termination of employment, other than payment of salary and benefits during the required notice period. Dr. Belzer’s agreement also provides for an annual bonus, payable within three months from the beginning of each calendar year, equal to up to two months’ salary based upon the successful achievement of objectives determined by our chief executive officer and in accordance with our compensation policy, subject to approval of our board of directors. As of March 15, 2019, Dr. Belzer held 2,550,000 options to purchase 2,083,333 ordinary shares at a weighted exercise price of NIS 0.61, of which 800,000 options are fully vested and 87,500 options will vest by August 31, 2019, and 562,500 options will vest over a period of three years from December 27, 2018, in equal parts at the end of every quarter thereafter and 1,100,000 options will vest over a period of four years, with a quarter of the options vesting on January 30, 2020, and the remaining options vesting in equal parts at the end of every quarter thereafter.
Dr. Nadav Orr
Dr. Orr has served as our vice president of research and development since September 2014. Dr. Orr is entitled to a monthly gross salary of NIS 40,000, and to social benefits, such as paid annual vacation days, severance pay, annual recreation allowance, manager’s insurance, sick leave, education fund and expenses reimbursement. In addition, we provide Dr. Orr with a leased company car and a mobile phone. Dr. Orr’s employment agreement is terminable by either us or Dr. Orr upon 90 days’ prior written notice. Dr. Orr’s employment agreement contains a non-compete obligation for a period of six months following termination of his employment and customary provisions regarding confidentiality of information and assignment of inventions. Dr. Orr’s employment agreement also provides for a grant of options to purchase up to 133,333 ordinary shares under the 2010 Plan, which will vest subject to certain conditions. The agreement does not provide for benefits upon the termination of employment, other than payment of salary and benefits during the required notice period. Dr. Orr’s agreement also provides for annual bonus equal to up to two months’ salary based upon successful achievement of objectives determined by our chief executive officer and in accordance with our compensation policy, within three months from the beginning of each calendar year and approval of our board of directors. As of March 15, 2019, Dr. Orr held 3,250,000 options to purchase 2,316,667 ordinary shares at a weighted average exercise price of NIS 0.67, of which 1,525,000 options are fully vested, 62,500 options will vest by May 19, 2019, and 562,500 options will vest over a period of the next three years from December 27, 2019, in equal parts at the end of every quarter thereafter and 1,100,000 options will vest over a period of four years, with a quarter of the options vesting on January 30, 2020, and the remaining options vesting in equal parts at the end of every quarter thereafter.
| C. | Board Practices |
Board of Directors
Under the Companies Law, the overseeing of the management of our business is vested in our board of directors. Our board of directors may exercise all powers and may take all actions that are not specifically granted to our shareholders or to management. Our officers are responsible for our day-to-day management and have individual responsibilities established by our board of directors and specified in their specific employment agreements. Our chief executive officer is appointed by, and serves at the discretion of, our board of directors, subject to the employment agreement that we have entered into with him. All other officers are appointed by our chief executive officer with the prior review of our board of directors and compensation committee, and are subject to the terms of any applicable employment agreements that we may enter into with them.
99
Under our articles of association, our board of directors must consist of at least three and not more than twelve directors, including at least two external directors. Our board of directors has decided to adopt an exemption that provides relief for Israeli companies whose shares are listed on certain stock exchanges outside of Israel (including the Nasdaq Capital Market) with no controlling shareholder, such as ourselves, from being required to appoint external directors so long as such companies satisfy the requirements of the foreign laws in the listing jurisdiction outside of Israel which apply to companies incorporated in such jurisdiction, in respect of the appointment of independent directors and the composition of the audit committee and compensation committee. An amendment to our articles of association that reflects such exemption is subject to shareholders approval, which is pending. Currently our board of directors consists of seven directors, including two external directors. Other than external directors, for whom special election requirements apply under the Companies Law, as detailed below, our articles of association provide that directors (other than external directors) are elected annually at the general meeting of our shareholders by a vote of the holders of a majority of the voting power present and voting, in person or by proxy, at that meeting.
We have three types of directors: independent directors, external directors (who are also independent in nature), and “regular” directors. For purposes of complying with the Nasdaq Listing Rules to list the Company’s ADSs on the Nasdaq Capital Market, our board of directors will be comprised of six independent directors (of which two are external directors).
Our board of directors has determined that with the exception of Adi Goldin, all of our directors are independent under such rules. The definition of “independent director” under the Nasdaq Listing Rules and “external director” under the Companies Law overlap to a significant degree such that we would generally expect the two directors serving as external directors to satisfy the requirements to be independent under the Nasdaq Listing Rules. The definition of external director under the Companies Law includes a set of statutory criteria that must be satisfied, including criteria whose aim is to ensure that there is no factor that would impair the ability of the external director to exercise independent judgment. The definition of independent director under Nasdaq Listing Rules specifies similar, if slightly less stringent, requirements in addition to the requirement that the board of directors consider any factor which would impair the ability of the independent director to exercise independent judgment. See “—External Directors” below for a description of the requirements under the Companies Law for a director to serve as an external director.
Under the Companies Law any shareholder holding at least 1% of our outstanding voting power may propose to nominate one or more persons for election as directors at a general meeting by delivering a written notice of such shareholder’s intent to make such nomination or nominations to our registered office. Each such notice must set forth all of the details and information as required to be provided by our amended and restated articles of association and regulations promulgated under the Companies law.
In addition, our articles of association allow our board of directors to appoint additional director or directors who shall remain in office until the next annual shareholders’ meeting, provided that the board of directors must consist of no more than 12 directors. In addition, our articles of association allow our board of directors to appoint alternate directors to fill vacancies on our board of directors, for a term of office equal to the remaining period of the term of office of the director(s) whose office(s) have been vacated.
Under the Companies Law, our board of directors must determine the minimum number of directors who are required to have accounting and financial expertise. See “—External Directors” below. In determining the number of directors required to have such expertise, our board of directors must consider, among other things, the type and size of the company and the scope and complexity of its operations. Our board of directors has determined that the minimum number of directors who are required to have accounting and financial expertise is one.
External Directors
Under the Companies Law, a public company is required to have at least two directors who qualify as external directors. Regulations promulgated under the Companies Law further provide relief for Israeli companies whose shares are listed on certain stock exchanges outside of Israel (including The Nasdaq Capital Market) with no controlling shareholder, such as ourselves, exempting such companies from being required to appoint external directors so long as such companies satisfy the requirements of the foreign laws in the listing jurisdiction outside of Israel which apply to companies incorporated in such jurisdiction, in respect of the appointment of independent directors and the composition of the audit committee and compensation committee. We presently have two external directors on our board of directors, but our board of directors has decided to adopt such exemption available to such dual-listed and foreign listed companies with no controlling shareholder. An amendment to our articles of association that reflects such exemption is subject to shareholders approval, which is pending. The appointment of our external directors was made by a resolution of the general meeting of our shareholders, and our external directors are Dr. Gili Hart and Dr. Elan Penn.
100
The Companies Law provides that external directors must be elected by a majority vote of the shares present and voting at a shareholders’ meeting, provided that either:
| ● | such majority includes at least a majority of the shares held by all shareholders who are not controlling shareholders and do not have a personal interest in the election of the external director (other than a personal interest not deriving from a relationship with a controlling shareholder) that are voted at the meeting, excluding abstentions, to which we refer as a disinterested majority; or |
| ● | the total number of shares voted against the election of the external director by non-controlling shareholders and by shareholders who do not have a personal interest in the election of the external director (other than a personal interest not deriving from a relationship with a controlling shareholder) does not exceed 2% of the aggregate voting rights in the company. |
Under the Companies Law, the term “controlling shareholder” means a shareholder with the ability to direct the activities of the company, other than by virtue of serving as an office holder. A shareholder is presumed to be a controlling shareholder if the shareholder holds 50% or more of the voting rights in a company or has the right to appoint more than half of the directors of the company or its general manager. For the purpose of approving transactions with controlling shareholders, a controlling shareholder is deemed to include any shareholder that holds 25% or more of the voting rights in a public company if no other shareholder holds more than 50% of the voting rights in the company. For purposes of determining the holding percentage stated above, two or more shareholders who have a personal interest in a transaction that is brought for the company’s approval are deemed as joint holders.
Under the Companies Law, the initial term of an external director is three years. Thereafter, an external director may be reelected to serve in that capacity for no more than two additional three-year terms, provided that either (i) his or her service for each such additional term is recommended by one or more shareholders holding at least 1% of the company’s voting rights and is approved at a shareholders’ meeting by a disinterested majority, where the total number of shares held by non-controlling, disinterested shareholders voting for such reelection exceeds 2% of the aggregate voting rights in the company, provided that the nominating shareholder, the external director, and certain of their related parties meet additional independence requirements; (ii) his or her service for each such additional term is recommended by the board of directors and is approved at a shareholders’ meeting by the same majority required for the initial election of an external director (as described above); or (iii) the external director has recommended that he or she be nominated for each such additional term and such nomination is approved at a shareholders’ meeting by the same majority and under the same criteria required as if he had been recommended by a shareholder.
The term of office for external directors for companies traded on certain foreign stock exchanges, including the Nasdaq Capital Market, may be further extended, in increments of additional three-year terms, provided that, in addition to reelection in such manner described above, (i) the audit committee and subsequently the board of directors of the company confirm that, in light of the external director’s expertise and special contribution to the work of the board of directors and its committees, the reelection for such additional period is beneficial to the company, and provided that (ii) the external director is reelected subject to the same shareholder vote requirements as if elected for the first time (as described above). Prior to the approval of the reelection of the external director at a general shareholders’ meeting, the company’s shareholders must be informed of the term previously served by him or her and of the reasons why the board of directors and audit committee recommended the extension of his or her term.
External directors may be removed from office by an extraordinary general meeting of shareholders called by the board of directors, which approves such dismissal by the same shareholder vote percentage required for their election or by a court, in each case only under limited circumstances, including ceasing to meet the statutory qualifications for appointment or violating their duty of loyalty to the company. If an external directorship becomes vacant and there are fewer than two external directors on the board of directors at the time, then the board of directors is required under the Companies Law to call a shareholders’ meeting as soon as possible to appoint a replacement external director.
101
Each committee of the board of directors that exercises the powers of the board of directors must include at least one external director. The audit committee and the compensation committee must include all external directors then serving on the board of directors and should be comprised of a majority of independent directors, the external directors must be the majority of the members of the compensation committee, and the audit and compensation committee’s chairman must be an external director. See “—Committees of the Board of Directors” below. Under the Companies Law, external directors of a company and all members of the compensation committee are prohibited from receiving, directly or indirectly, any compensation for their services, other than for their services as external directors pursuant to the Companies Law and the regulations promulgated thereunder. Compensation of an external director is determined prior to his or her appointment and may not be changed during his or her term subject to certain exceptions. Under the regulations pursuant to the Companies Law, certain exemptions and reliefs are granted to companies which securities are traded outside of Israel. We may use those exemptions and reliefs in the future.
The Companies Law provides that a person is not qualified to serve as an external director if (i) the person is a relative of a controlling shareholder of the company or (ii) if that person or his or her relative, partner, employer, another person to whom he or she was directly or indirectly subject, or any entity under the person’s control, has or had, during the two years preceding the date of appointment as an external director: (a) any affiliation or other disqualifying relationship with the company, with any person or entity controlling the company or a relative of such person, or with any entity controlled by or under common control with the company; or (b) in the case of a company with no shareholder holding 25% or more of its voting rights, had, at the date of appointment as external director, any affiliation or other disqualifying relationship with a person then serving as chairman of the board or chief executive officer, a holder of 5% or more of the issued share capital or voting power in the company, or the most senior financial officer.
The term “relative” is defined under the Companies Law as a spouse, sibling, parent, grandparent, or descendant; spouse’s sibling, parent, or descendant; and the spouse of each of the foregoing persons. Under the Companies Law, the term “affiliation” and the similar types of prohibited relationships include (subject to certain exceptions):
| ● | an employment relationship; |
| ● | a business or professional relationship even if not maintained on a regular basis (excluding insignificant relationships); |
| ● | control; and |
| ● | service as an office holder, excluding service as a director in a private company prior to the initial public offering of its shares if such director were appointed as a director of the private company in order to serve as an external director following the initial public offering. |
The term office holder is defined under the Companies Law as the general manager, chief executive officer, chief business manager, deputy general manager, vice general manager, any other person assuming the responsibilities of any of these positions regardless of that person’s title, and a director, or a manager directly subordinate to the general manager.
In general, the external directors must be of Israeli residency (unless the company on which he or she serves had offered shares (or bonds) to the public outside of Israel or are registered on a stock exchange outside of Israel) and must possess the minimal criteria required for the directorship of a “regular” director. In addition, no person may serve as an external director if that person’s position or professional or other activities create, or may create, a conflict of interest with that person’s responsibilities as a director or otherwise interfere with that person’s ability to serve as an external director or if the person is an employee of the ISA or of an Israeli stock exchange. A person may furthermore not continue to serve as an external director if he or she received direct or indirect compensation from the company including amounts paid pursuant to indemnification or exculpation contracts or commitments and insurance coverage for his or her service as an external director, other than as permitted by the Companies Law and the regulations promulgated thereunder.
102
For a period of two years from the date that an external director of a company ceases to act in such capacity, the company in which such external director served, and its controlling shareholder or any entity under control of such controlling shareholder may not, directly or indirectly, grant such former external director or his or her spouse or child, any benefit, including via (i) the appointment of such former director or his or her spouse or his child as an officer in the company or in an entity controlled by the company’s controlling shareholder, (ii) the employment of such former external director and (iii) the engagement, directly or indirectly, of such former external director as a provider of professional services for compensation, including via an entity under his or her control. With respect to a relative who is not a spouse or a child, such limitations shall only apply for one year from the date such external director ceased to be engaged in such capacity.
If, at the time at which an external director is appointed, all members of the board of directors, who are not controlling shareholders or relatives of controlling shareholders of the company, are of the same gender, the external director to be appointed must be of the other gender. A director of one company may not be appointed as an external director of another company if a director of the other company is acting as an external director of the first company at such time.
According to the Companies Law, a person may be appointed as an external director only if he or she has professional qualifications or if he or she has accounting and financial expertise (each, as defined below). In addition, at least one of the external directors must be determined by our board of directors to have accounting and financial expertise.
According to regulations promulgated under the Companies Law, a director with accounting and financial expertise is a director who, due to his or her education, experience, and skills, possesses an expertise in, and an understanding of, financial and accounting matters and financial statements, such that he or she is able to understand the financial statements of the company and initiate a discussion about the presentation of financial data. A director is deemed to have professional qualifications if he or she has: (i) an academic degree in economics, business management, accounting, law, or public administration; (ii) an academic degree, or has completed other higher education, in the primary field of business of the company or a field which is relevant to his or her position in the company; or (iii) at least five years of experience serving in one of the following capacities, or at least five years cumulative experience serving in two or more of the following capacities: (a) a senior business management position in a company with a significant volume of business; (b) a senior position in a company’s primary field of business; or (c) a senior position in public administration or service. The board of directors is charged with determining whether a director possesses financial and accounting expertise or professional qualifications.
Role of Board of Directors in Risk Oversight Process
Risk assessment and oversight are an integral part of our governance and management processes. Our board of directors encourages management to promote a culture that incorporates risk management into our corporate strategy and day-to-day business operations. Management discusses strategic and operational risks at regular management meetings and conducts specific strategic planning and review sessions during the year that include a focused discussion and analysis of the risks facing us. Throughout the year, senior management reviews these risks with the board of directors at regular board meetings as part of management presentations that focus on particular business functions, operations or strategies, and presents the steps taken by management to mitigate or eliminate such risks.
Leadership Structure of the Board of Directors
In accordance with the Companies Law and our articles of association, our board of directors is required to appoint one of its members to serve as chairman of the board of directors. Our board of directors has appointed Jonathan M.N. Rigby to serve as chairman of the board of directors.
Committees of the Board of Directors
Currently, our board of directors has four permanent committees: an audit committee, a compensation committee, financial statements committee and a nominating and corporate governance committee. The first two committees are mandatory and regulated under the Companies Law provisions. A nominating and corporate governance committee has been constituted.
103
Audit Committee
Under the Companies Law, we are required to appoint an audit committee. The audit committee of a public company must be comprised of at least three directors, including all of the external directors, one of whom must serve as chairman of the committee. The audit committee may not include the chairman of the board, any director employed by or otherwise providing services on a regular basis to the company, to a controlling shareholder, or to any entity controlled by a controlling shareholder, any director who derives most of his or her income from a controlling shareholder, nor a controlling shareholder or a relative thereof.
In addition, under the Companies Law, the audit committee of a publicly traded company must consist of a majority of independent directors. In general, an “independent director” under the Companies Law is defined as either an external director or as a director who meets the following criteria:
| ● | he or she meets the qualifications for being appointed as an external director and the audit committee has approved that he or she meets such qualifications, except for the requirement (i) that the director be an Israeli resident (which does not apply to companies such as ours whose securities have been offered outside of Israel to date or are listed outside of Israel) and (ii) for accounting and financial expertise or professional qualifications; and |
| ● | he or she has not served as a director of the company for a period exceeding nine consecutive years. For this purpose, a break of less than two years in the service shall not be deemed to interrupt the continuation of the service. |
Under the Nasdaq Listing Rules, we are required to maintain an audit committee consisting of at least three independent directors, all of whom are financially literate and at least one of whom has accounting or related financial management expertise.
Recent amendments to regulations promulgated under the Companies Law exempt Israeli companies whose shares are listed on certain stock exchanges outside of Israel (including The Nasdaq Capital Market) with no controlling shareholder, such as ourselves, from certain Companies Law provisions with respect to the composition of the audit committee and the quorum and majority requirements at its meetings, so long as such companies satisfy the requirements of the foreign laws in the listing jurisdiction outside of Israel which apply to companies incorporated in such jurisdiction in respect of the appointment of independent directors and the composition of the audit committee and compensation committee. Presently, we have an audit committee in place which composition complies with the listing requirements of the Companies Law, although we may elect in the future to rely on such exemption available to dual-listed companies with no controlling shareholder.
Our audit committee consists of Dr. Gili Hart, Dr. Elan Penn and Scott Burell. Dr. Penn and Mr. Burell possess accounting and financial expertise and are both audit committee financial experts as defined by the SEC rules, and all of the members of our audit committee have the requisite financial literacy as defined by the Nasdaq Listing Rules. All audit committee members are “independent” as such term is defined in Rule 10A3(b)(1) under the Exchange Act and under the Nasdaq Listing Rules.
Our board of directors has adopted an audit committee charter setting forth the responsibilities of the audit committee consistent with the rules of the SEC and the Nasdaq Listing Rules as well as the requirements for such committee under the Companies Law, including the following:
| ● | oversight of our independent registered public accounting firm and recommending the engagement, compensation or termination of engagement of our independent registered public accounting firm to the board of directors in accordance with Israeli law; |
| ● | recommending the engagement or termination of the person filling the office of our internal auditor; and |
| ● | recommending the terms of audit and non-audit services provided by the independent registered public accounting firm for pre-approval by our board of directors. |
104
Our audit committee provides assistance to our board of directors in fulfilling its legal and fiduciary obligations in matters involving our accounting, auditing, financial reporting, internal control, and legal compliance functions by pre-approving the services performed by our independent accountants and reviewing their reports regarding our accounting practices and systems of internal control over financial reporting. Our audit committee also oversees the audit efforts of our independent accountants and takes those actions that it deems necessary to satisfy itself that the accountants are independent of management.
Under the Companies Law, our audit committee is mainly responsible for:
| ● | determining whether there are deficiencies in our business management practices, including in consultation with our internal auditor or the independent auditor, and making recommendations to the board of directors to improve such practices; |
| ● | determining whether certain acts of an office holder not in accordance with his or her fiduciary duty owed to the Company are extraordinary or material and to approve such acts and certain related party transactions (including transactions in which an office holder has a personal interest) and whether such transaction is extraordinary or material under the Companies Law (see “—Approval of Related Party Transactions Under Israeli Law” below); |
| ● | determining procedures for a competitive process, or other procedures, before approving related party transactions with controlling shareholders, even if such transactions are deemed by the audit committee not to be extraordinary transactions. This process is to be supervised by the audit committee, or any person authorized for such supervision, or via any other method approved by the audit committee; |
| ● | determining the approval process for transactions that are not negligible, as well as determine which types of transactions would require the approval of the audit committee. Non-negligible transactions are defined as related party transactions with a controlling shareholder, or in which the controlling shareholder has a personal interest, even if they are deemed by the audit committee not to be extraordinary transactions but which have also been classified by the audit committee as non-negligible transactions; |
| ● | where the board of directors approves the work plan of the internal auditor, to examine such work plan before its submission to the board and propose amendments thereto; |
| ● | examining our internal controls and internal auditor’s performance, including whether the internal auditor has sufficient resources and tools to dispose of its responsibilities; |
| ● | examining the scope of our auditor’s work and compensation and submitting a recommendation with respect thereto to our board of directors or shareholders, depending on which of them is considering the appointment of our auditor; and |
| ● | establishing procedures for the handling of employees’ complaints as to deficiencies in the management of our business and the protection to be provided to such employees. |
Our audit committee may not approve any actions requiring its approval (see “—Approval of Related Party Transactions Under Israeli Law” below), unless at the time of approval a majority of the committee’s members are present, which majority consists of independent directors including at least one external director.
105
Compensation Committee
Our compensation committee consists of Dr. Abraham Havron, Dr. Gili Hart and Dr. Elan Penn.
Under the Companies Law, the board of directors of a public company must appoint a compensation committee. Subject to certain exceptions compensation committee must be comprised of at least three directors, including all of the external directors, which shall be a majority of the members of the compensation committee and one of whom must serve as chairman of the committee.
Each compensation committee member who is not an external director must be a director whose compensation is equivalent to the compensation that may be paid to an external director. The compensation committee is subject to the same Companies Law restrictions as the audit committee as to who may not be a member of the committee. According to the Companies Law, our audit committee may also act as compensation committee.
Recent amendments to regulations promulgated under the Companies Law exempt Israeli companies whose shares are listed on certain stock exchanges outside of Israel (including the Nasdaq Capital Market) with no controlling shareholder, such as ourselves, from the Companies Law requirements to appoint a compensation committee or of its composition, so long as such companies satisfy the requirements of the foreign laws in the listing jurisdiction outside of Israel which apply to companies incorporated in such jurisdiction in respect of the appointment of independent directors and the composition of the audit committee and compensation committee. Presently, we have a compensation committee in place which composition complies with the requirements of the Companies Law, although we may elect in the future to rely on such exemption available to dual-listed companies with no controlling shareholder.
The duties of the compensation committee include the recommendation to the company’s board of directors of a policy regarding the terms of engagement of office holders, to which we refer as a compensation policy, and to examine the necessity of updating the compensation policy. That policy must be adopted by the company’s board of directors, after considering the recommendations of the compensation committee, and must be approved by the company’s shareholders, which approval requires a special majority. For this purpose, a “special majority” approval requires shareholder approval by a majority vote of the shares present and voting at a meeting of shareholders called for such purpose, provided that either: (i) such majority includes at least a majority of the shares held by all shareholders who are not controlling shareholders and do not have a personal interest in such compensation arrangement; or (ii) the total number of shares of non-controlling shareholders and shareholders who do not have a personal interest in the compensation arrangement and who vote against the arrangement does not exceed 2% of the company’s aggregate voting rights. Under special circumstances, the board of directors may approve the compensation policy despite the objection of the shareholders on the condition that the compensation committee (or the audit committee acting in lieu of a compensation committee pursuant to the Companies Law) and then the board of directors decide, on the basis of detailed arguments and after discussing again the compensation policy, that approval of the compensation policy, despite the objection of the meeting of shareholders, is for the benefit of the company. Our current compensation policy was approved by our shareholders on March 1, 2018 and will be in effect for a period of three years from the date of approval. We have adopted a new compensation policy which remains subject to shareholder approval. The compensation policy does not, by nature, grant any rights to our directors or officers. The compensation policy includes both long-term and short-term compensation elements and is to be reviewed from time to time by our compensation committee and our board of directors, according to the requirements of the Companies Law.
Our compensation policy serves as the basis for decisions concerning the financial terms of employment or engagement of office holders, including exculpation, insurance, indemnification or any monetary payment or obligation of payment with respect to employment or engagement. According to the Companies Law, the compensation policy must be approved (or reapproved) not longer than every three years and relate to certain factors, including advancement of the company’s objectives, the company’s business plan and its long-term strategy, and creation of appropriate incentives for office holders. It must also consider, among other things, the company’s risk management, size, and nature of its operations. The compensation policy must furthermore consider the following additional factors:
| ● | the knowledge, skills, expertise, and accomplishments of the relevant office holder; |
| ● | the office holder’s roles and responsibilities and prior compensation agreements with him or her; |
106
| ● | the ratio between the terms offered and the average compensation of the other employees of the company, including those employed through manpower companies, and in particular the ratio between the average wage and the median salary of such employees; |
| ● | the impact of disparities in salary upon work relationships in the company; |
| ● | the possibility of reducing variable compensation at the discretion of the board of directors; |
| ● | the possibility of setting a limit on the exercise value of non-cash variable equity-based compensation; and |
| ● | as to severance compensation, the period of service of the office holder, the terms of his or her compensation during such service period, the company’s performance during that period of service, the person’s contributions towards the company’s achievement of its goals and the maximization of its profits, and the circumstances under which the person is leaving the company. |
The compensation policy must also include the following principles:
| ● | the linkage between variable compensation and long-term performance and measurable criteria; however, in certain circumstances, we may grant up to three monthly salaries per year of unmeasurable criteria for an office holder who is not our chief executive officer. |
| ● | the ratio between variable and fixed compensation, and the ceiling for the value of variable compensation at the time of the payment (or with respect to variable equity compensation that is not paid for in cash, a ceiling for their value on the grant date); |
| ● | the conditions under which an office holder would be required to repay compensation paid to him or her if it was later shown that the data upon which such compensation was based was inaccurate and was required to be restated in the company’s financial statements; |
| ● | the minimum holding or vesting period for variable, equity-based compensation with a view to long-term incentives; and |
| ● | maximum limits for severance compensation. |
Our board of directors has adopted a compensation committee charter setting forth the responsibilities of the committee, which include:
| ● | the responsibilities set forth in the compensation policy; |
| ● | reviewing and approving the granting of options and other incentive awards to the extent such authority is delegated by our board of directors; and |
| ● | reviewing, evaluating, and making recommendations regarding the compensation and benefits for our non-employee directors. |
Financial Statements Committee
Our financial statements committee, which complies with the Israeli Companies Regulations (Provisions and Conditions Regarding the Financial Statements’ Authorization Process), 2010, is responsible for considering and making recommendations to the board of directors on our financial statements. Prior to the approval of our financial statements by our board of directors, the financial statements committee reviews and discusses the financial statements and presents its recommendations with respect to the financial statements to the board of directors. Our financial statements committee currently consists of the members of our audit committee: Dr. Gili Hart, Mr. Scott R. Burell and Dr. Elan Penn.
107
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee consists of Dr. Gili Hart, Dr. Abraham Havron, and Dr. Elan Penn. Each of the members of our nominating and corporate governance committee is independent under the listing requirements of the Nasdaq Capital Market.
Our board of directors has adopted a nominating and governance committee charter setting forth the responsibilities of the nominating and governance committee, which include:
| ● | overseeing and assisting our board in reviewing and recommending nominees for election as directors; |
| ● | assessing the performance of the members of our board; and |
| ● | establishing and maintaining effective corporate governance policies and practices, including, but not limited to, developing and recommending to our board a set of corporate governance guidelines applicable to our company. |
Internal Auditor
Under the Companies Law, the board of directors of a public company must appoint an internal auditor based on the recommendation of the audit committee. The role of the internal auditor is to examine, among other things, our compliance with applicable law and orderly business procedures. The audit committee is required to oversee the activities and to assess the performance of the internal auditor as well as to review the internal auditor’s work plan.
An internal auditor may not be:
| ● | a person (or a relative of a person) who holds more than 5% of the company’s outstanding shares or voting rights; |
| ● | a person (or a relative of a person) who has the power to appoint a director or the general manager of the company; |
| ● | an office holder or director (or a relative of an officer or director) of the company; or |
| ● | a member of the company’s independent accounting firm, or anyone on its behalf. |
Ms. Dana Gottesman Erlich, has been serving as our Internal Auditor since November 2013. Ms. Gottesman is a CPA, CIA, MA, Partner in the Risk Advisory Services (RAS) Group at the accounting firm of BDO Ziv Haft. Ms. Gottesman has more than 10 years of experience in the provision of internal audit and risk management consulting services to public and private companies, government agencies, municipalities, non-profit organizations, and more. Ms. Gottesman specializes in the analysis and specification of work procedures and their assimilation in the organization, the internal audit of work procedures in different organizations, including the performance of risk surveys and fraud and embezzlement surveys. Ms. Gottesman holds a BA in Accounting and Business Administration and an MA in Internal Audit and Public Administration. Ms. Gottesman’s nomination satisfies the requirements of the Companies Law.
Approval of Related Party Transactions under Israeli Law
Fiduciary Duties of Directors and Officers
The Companies Law imposes a duty of care and a fiduciary duty on all office holders of a company. Each person listed in the table under “Management—Senior Management and Directors” is an office holder under the Companies Law.
108
The duty of care requires an office holder to act with the degree of proficiency with which a reasonable office holder in the same position would have acted under the same circumstances. The fiduciary duty requires that an office holder act in good faith and in the best interests of the company.
The duty of care includes a duty to use reasonable means to obtain:
| ● | information on the advisability of a given action brought for his or her approval or performed by virtue of his or her position; and |
| ● | all other important information pertaining to these actions. |
The fiduciary duty includes a duty to:
| ● | refrain from any act involving a conflict of interest between the performance of his or her duties to the company and his or her other duties or personal affairs; |
| ● | refrain from any activity that is competitive with the company; |
| ● | refrain from exploiting any business opportunity of the company to receive a personal gain for himself or herself or others; and |
| ● | disclose to the company any information or documents relating to the company’s affairs which the office holder received as a result of his or her position as an office holder. |
Disclosure of Personal Interests of an Office Holder and Approval of Certain Transactions
The Companies Law requires that an office holder promptly disclose to the company any personal interest that he or she may be aware of and all related material information or documents concerning any existing or proposed transaction by the company. An interested office holder’s disclosure must be made promptly and, in any event, no later than the first meeting of the board of directors at which the transaction is considered. An office holder is not obliged to disclose a personal interest if it derives solely from the personal interest of his or her relative in a transaction that is not considered as an extraordinary transaction.
A “personal interest” is defined under the Companies Law to include a personal interest of any person in an act or transaction of a company, including the personal interest of such person’s relative or of a corporate body in which such person or a relative of such person is a 5% or greater shareholder, director, or general manager or in which he or she has the right to appoint at least one director or the general manager, but excluding a personal interest solely stemming from one’s ownership of shares in the company.
A personal interest furthermore includes the personal interest of a person for whom the office holder holds a voting proxy or the personal interest of the office holder with respect to his or her vote on behalf of a person for whom he or she holds a proxy even if such shareholder has no personal interest in the matter. An office holder is not, however, obliged to disclose a personal interest if it derives solely from the personal interest of his or her relative in a transaction that is not considered an extraordinary transaction.
Under the Companies Law, an extraordinary transaction is defined as any of the following:
| ● | a transaction other than in the ordinary course of business; |
| ● | a transaction that is not on market terms; or |
| ● | a transaction that may have a material impact on the company’s profitability, assets, or liabilities. |
109
If it is determined that an office holder has a personal interest in a transaction which is not an extraordinary transaction, approval by the board of directors is required for such transaction, unless the company’s articles of association provide for a different method of approval. An extraordinary transaction in which an office holder has a personal interest requires approval first by the company’s audit committee and subsequently by the board of directors. In general, the compensation of, or an undertaking to indemnify or insure, an office holder who is not a director requires approval first by the company’s compensation committee, then by the company’s board of directors, and, if such compensation arrangement or an undertaking to indemnify or insure is inconsistent with the company’s stated compensation policy or if the office holder is the chief executive officer (apart from a number of specific exceptions), then such arrangement is subject to a special majority approval. Arrangements regarding the compensation, exculpation, indemnification, or insurance of a director require the approval of the compensation committee, board of directors, and shareholders by ordinary majority, in that order, and under certain circumstances, a special majority approval.
Generally, a person who has a personal interest in a matter which is considered at a meeting of the board of directors or the audit committee may not be present at such a meeting or vote on that matter unless the chairman of the relevant committee or board of directors (as applicable) determines that he or she should be present in order to present the transaction that is subject to approval. If a majority of the members of the audit committee or the board of directors (as applicable) has a personal interest in the approval of a transaction, then all directors may participate in discussions of the audit committee or the board of directors (as applicable) on such transaction and the voting on approval thereof, but shareholder approval is also required for such transaction.
Disclosure of Personal Interests of Controlling Shareholders and Approval of Certain Transactions
Under Israeli Law, the term “controlling shareholder” means a shareholder with the ability to direct the activities of our company, other than by virtue of being an executive officer or director. A shareholder is presumed to be a controlling shareholder if the shareholder holds 50% or more of the voting rights in a company or has the right to appoint at least half of the directors of the company or its general manager. For the purpose of approving transactions with controlling shareholders, a controlling shareholder is deemed to include any shareholder that holds 25% or more of the voting rights in a public company if no other shareholder holds more than 50% of the voting rights in the company. For purposes of determining the holding percentage stated above, two or more shareholders who have a personal interest in a transaction that is brought for the company’s approval are deemed as joint holders.
Pursuant to Israeli law, the disclosure requirements regarding personal interests that apply to directors and executive officers also apply to a controlling shareholder of a public company. See “—External Directors” above for a definition of controlling shareholder. In the context of a transaction involving a shareholder of the company, a controlling shareholder also includes a shareholder who holds 25% or more of the voting rights in the company if no other shareholder holds more than 50% of the voting rights in the company. For this purpose, the holdings of all shareholders who have a personal interest in the same transaction will be aggregated. The approval of the audit committee or compensation committee, the board of directors, and a special majority, in that order, is required for: (i) extraordinary transactions with a controlling shareholder or in which a controlling shareholder has a personal interest; (ii) the engagement with a controlling shareholder or his or her relative, directly or indirectly, for the provision of services to the company; (iii) the terms of engagement and compensation of a controlling shareholder or his or her relative who is not an office holder; or (iv) the employment of a controlling shareholder or his or her relative by the company, other than as an office holder. For this purpose, a “special majority” approval requires shareholder approval by a majority vote of the shares present and voting at a meeting of shareholders called for such purpose, provided that either: (a) such majority includes at least a majority of the shares held by all shareholders who do not have a personal interest in such compensation arrangement; or (b) the total number of shares of non-controlling shareholders and shareholders who do not have a personal interest in the compensation arrangement and who vote against the arrangement does not exceed 2% of the company’s aggregate voting rights.
To the extent that any such transaction with a controlling shareholder is for a period extending beyond three years, approval is required once every three years, unless, with respect to certain transactions, the audit committee determines that the duration of the transaction is reasonable given the circumstances related thereto.
Arrangements regarding the compensation, exculpation, indemnification, or insurance of a controlling shareholder in his or her capacity as an office holder require the approval of the compensation committee and board of directors, and, in general, approval by a special majority of shareholders.
110
Pursuant to regulations promulgated under the Companies Law, certain transactions with a controlling shareholder or his or her relative, or with directors, that would otherwise require approval of a company’s shareholders may be exempt from shareholder approval upon certain determinations of the audit committee or compensation committee and board of directors.
Shareholders’ Duties
Under the Companies Law, a shareholder has a duty to act in good faith and in a customary manner toward the company and other shareholders and to refrain from abusing his or her power in the company, including, among other things, in voting at general meetings of shareholders and class meetings of shareholders with respect the following matters:
| ● | an amendment of the articles of association or memorandum of association of the company; |
| ● | an increase in the company’s authorized share capital; |
| ● | a merger; or |
| ● | the approval of related party transactions and acts of office holders that require shareholder approval. |
A shareholder also has a general duty to refrain from discriminating against other shareholders. In addition, certain shareholders have a duty of fairness toward the company. These shareholders include any controlling shareholder, any shareholder who knows that he or she has the power to determine the outcome of a shareholder vote and any shareholder who has the power to appoint or to prevent the appointment of an office holder of the company or other power. The Companies Law does not define the substance of the duty of fairness, except to state that the remedies generally available upon a breach of contract will also apply in the event of a breach of the duty to act with fairness.
Exculpation, Insurance and Indemnification of Directors and Officers
Under the Companies Law, a company may not exculpate an office holder from liability for a breach of the duty of loyalty. An Israeli company may exculpate an office holder in advance from liability to the company, in whole or in part, for damages caused to the company as a result of a breach of duty of care but only if a provision authorizing such exculpation is included in its articles of association. Our articles of association include such a provision. A company may not exculpate a director from liability arising out of a prohibited dividend or distribution to shareholders.
Under the Companies Law and the Israeli Securities Law, an Israeli company may indemnify an office holder with respect to the following liabilities and expenses incurred for acts performed as an office holder, either in advance of an event or following an event, provided a provision authorizing such indemnification is contained in its articles of association:
| ● | financial liability imposed on him or her in favor of another person pursuant to a judgment, including a settlement or arbitrator’s award approved by a court. However, if an undertaking to indemnify an office holder with respect to such liability is provided in advance, then such an undertaking must be limited to events which, in the opinion of the board of directors, can be foreseen based on the company’s activities when the undertaking to indemnify is given, and to an amount or according to criteria determined by the board of directors as reasonable under the circumstances, and such undertaking must detail the abovementioned foreseen events and amount or criteria; |
| ● | reasonable litigation expenses, including attorneys’ fees, incurred by the office holder: (i) as a result of an investigation or proceeding instituted against him or her by an authority authorized to conduct such investigation or proceeding, provided that (a) no indictment was filed against such office holder as a result of such investigation or proceeding and (b) no financial liability was imposed upon him or her as a substitute for the criminal proceeding as a result of such investigation or proceeding or, if such financial liability was imposed, it was imposed with respect to an offense that does not require proof of criminal intent; and (ii) in connection with a monetary sanction; |
111
| ● | expenses associated with an administrative procedure, as defined in the Israeli Securities Law, conducted regarding an office holder, including reasonable litigation expenses and reasonable attorneys’ fees; and |
| ● | reasonable litigation expenses, including attorneys’ fees, incurred by the office holder or imposed by a court in proceedings instituted against him or her by the company, on its behalf, or by a third party or in connection with criminal proceedings in which the office holder was acquitted or as a result of a conviction for an offense that does not require proof of criminal intent. |
Under the Companies Law and the Israeli Securities Law, a company may insure an office holder against the following liabilities incurred for acts performed as an office holder if, and to the extent, provided in the company’s articles of association:
| ● | a breach of duty of care to the company or to a third party, including a breach arising out of the negligent conduct of the office holder; |
| ● | a breach of fiduciary duty to the company, to the extent that the office holder acted in good faith and had a reasonable basis to believe that the act would not prejudice the company; |
| ● | a monetary liability imposed on the office holder in favor of a third party; and |
| ● | expenses incurred by an office holder in connection with an administrative procedure, including reasonable litigation expenses and reasonable attorneys’ fees. |
Under the Companies Law, a company may not indemnify or insure an office holder against any of the following:
| ● | a breach of fiduciary duty, except for indemnification and insurance for a breach of the fiduciary duty to the company and to the extent that the office holder acted in good faith and had a reasonable basis to believe that the act would not prejudice the company; |
| ● | a breach of duty of care committed intentionally or recklessly, excluding a breach arising out of the negligent conduct of the office holder; |
| ● | an act or omission committed with intent to derive illegal personal benefit; or |
| ● | a fine or forfeit levied against the office holder. |
Under the Companies Law, exculpation, indemnification, and insurance of office holders in a public company must be approved by the compensation committee and the board of directors and, with respect to certain office holders or under certain circumstances, by the shareholders.
Our articles of association and compensation policy allow us to exculpate, indemnify, and insure our office holders according to applicable law.
As of the date of this Annual Report on Form 20-F, no claims for directors’ and officers’ liability insurance have been filed under this policy and we are not aware of any pending or threatened litigation or proceeding involving any of our directors or officers in which indemnification is sought.
We have obtained directors’ and officers’ liability insurance for the benefit of our office holders and intend to continue to maintain such coverage and pay all premiums thereunder to the fullest extent permitted by the Companies Law. In addition, we have entered into agreements with each of our current office holders undertaking to indemnify them to the fullest extent permitted by the Companies Law and our articles of association, to the extent that these liabilities are not covered by insurance.
112
In the opinion of the Securities and Exchange Commission, indemnification of directors and office holders for liabilities arising under the Securities Act, however, is against public policy and therefore unenforceable.
There is no pending litigation or proceeding against any of our directors or officers as to which indemnification is being sought, nor are we aware of any pending or threatened litigation that may result in claims for indemnification by any director or officer.
| D. | Employees. |
See “Item 4.B. Business Overview―Employees.”
| E. | Share Ownership. |
See “Item 7.A. Major Shareholders” below.
Share Incentive Plan
In May 2010, we adopted the 2010 Plan, an option plan for employees and senior officers, and as part of the acquisition of CollPlant Ltd., all of the options under the Employee Share Ownership and Option Plan (2004) of CollPlant Ltd. were substituted with and assumed by options under our 2010 Plan, while any restriction periods under Sections 102(b)(2) and 102(b)(3) of the Israeli Income Tax Ordinance, or the Ordinance, were calculated as of their original grant date. The 2010 Plan allows us to grant options to purchase our ordinary shares to our officers, employees, and consultants. The 2010 Plan is intended to enhance our ability to attract and retain desirable individuals by increasing their ownership interests in us. As of March 15, 2019, our employees, officers, and consultants hold an aggregate of 55,919,792 options to purchase 35,613,931 ordinary shares under the 2010 Plan. In addition, on January 30, 2019, our board of directors approved the grant of 17,019,500 options to purchase 17,019,500 ordinary shares subject to shareholders approval. As of March 15, 2019, 7,464,183 options to purchase an aggregate of 2,488,061 ordinary shares had been exercised and transferred to the beneficial holders. The 2010 Plan is designed to reflect the provisions of the Israeli Income Tax Ordinance, or the Ordinance, mainly Sections 102 and 3(i), which affords certain tax advantages to Israeli employees, officers, and directors that are granted options in accordance with its terms. Section 102 of the Ordinance allows employees, directors, and officers, who are not controlling shareholders and who are Israeli residents, to receive favorable tax treatment for compensation in the form of shares or options. Section 102 of the Ordinance includes two alternatives for tax treatment involving the issuance of options or shares to a trustee for the benefit of the grantees and also includes an additional alternative for the issuance of options or shares directly to the grantee. Sections 102(b)(2) and 102(b)(3) of the Ordinance, which provide the most favorable tax treatment for grantees, permit the issuance to a trustee under the “capital gains track.” In order to comply with the terms of the capital gains track, all options granted under a specific plan and subject to the provisions of Section 102 of the Ordinance, as well as the shares issued upon exercise of such options and other shares received following any realization of rights with respect to such options, such as share dividends and share splits, must be registered in the name of a trustee selected by the board of directors and held in trust for the benefit of the relevant employee, director, or officer. The trustee may not release these options or shares to the relevant grantee before the second anniversary of the registration of the options in the name of the trustee. However, under this track, our ability to deduct an expense with respect to the issuance of the options or shares might be limited. Section 3(i) of the Ordinance does not provide for similar tax benefits.
The plans may be administered by our board of directors either directly or upon the recommendation of a committee appointed by our board of directors.
The compensation committee recommends to the board of directors, and the board of directors determines or approves, the eligible individuals who receive options under the plan, the number of ordinary shares covered by those options, the terms under which such options may be exercised, and other terms and conditions of the options, all in accordance with the provisions of the plans. Option holders may not transfer their options except in the event of death or transfer to an Administrator in accordance with law in the event of the absence of legal competency. Our compensation committee or board of directors may, at any time, amend or terminate each of the plans; however, any amendment or termination may not adversely affect any options or shares granted under such plan prior to such action.
113
The option exercise price is determined by the compensation committee, following the approval of the board of directors, and specified in each option award agreement. In general, and according to our compensation policy, the option exercise price is the market value of the shares on the date of grant in accordance to the ADS market value traded on the Nasdaq Capital Market.
Awards under the 2010 Plan may be granted until 2020, 10 years from the date on which the 2010 Plan was approved by our board of directors.
Options granted under the 2010 Plan generally vest over four years commencing on the date of grant such that 25% vest on the first anniversary of the date of grant and an additional 6.25% vest at the end of each subsequent three-month period thereafter for 36 months and some every calendar year, unless otherwise provided in a specific allocation agreement.
Options, other than certain incentive share options, that are not exercised within 10 years from the grant date expire, unless otherwise determined by our board of directors. Except as otherwise determined by the board of directors or as set forth in an individual’s award agreement, in the event of termination of employment or services for reasons of disability, death, or retirement, the grantee, or in the case of death, his or her legal successor, may exercise options that have vested prior to termination within a period of one year from the date of disability, death, or retirement. If we terminate a grantee’s employment or service for cause, all of the grantee’s unvested options will expire on the date of termination, yet options which by that date the offeree’s eligibility to exercise has already been formed shall remain exercisable. If a grantee’s employment or service is terminated for any other reason, the grantee may exercise his or her vested options within 90 days of the date of termination. Any expired or unvested options return to the pool for reissuance.
In the event of (i) a sale of all or substantially all of our assets or (ii) our consolidation or merger in which we are not the ongoing or surviving corporation, then, and unless otherwise determined in the agreement or by the board, we shall be entitled to determine that all of the outstanding unexercised options held by or for the benefit of any grantee shall be assumed or substituted for an appropriate number of options of the successor company, provided that the aggregate amount of the exercise price for such options shall be equal to the aggregate amount of the exercise price of our unexercised options held by each grantee at such time. With respect to the grants that were made since October 2017, the above acceleration provision was amended in a manner that the options’ vesting is fully accelerated upon the occurrence of a M&A Transaction or Reorganization: (1) “M&A Transaction” shall mean a “merger” as such term or term of similar nature is defined in the Israeli Companies Law of 1999, as well as (i) a sale of 50% or more of the assets of the Company and its subsidiaries taken as a whole, or the sale or disposition (whether by merger or otherwise) of one or more subsidiaries of the Company if more than 50% of the assets of the Company and its subsidiaries taken as a whole are held by such subsidiary or subsidiaries; (ii) a sale of all or more than 50% of the shares of the share capital of the Company whether by a single transaction or a series of related transactions which occur either over a period of 12 months or within the scope of the same acquisition agreement; (iii) an issuance of shares of the Company, whether by a single transaction or a series of related transactions which occur either over a period of 12 months or within the scope of the same acquisition agreement, that results in the offeree holding more than 50% of the share capital of the Company; or (iv) a merger, consolidation or like transaction of the Company with or into another corporation including a reverse triangular merger, but excluding a merger which falls within the definition of Reorganization; and/or (2) “Reorganization” shall mean any re-domestication of the Company, share flip, creation of a holding Company for the Company which will hold all, or 50% or more, of the shares of the Company or any other transaction involving the Company in which the ordinary shares of the Company outstanding immediately prior to such transaction continue to represent, or are converted into or exchanged for shares that represent, immediately following such transaction, at least a majority, by voting power, of the share capital of the surviving, acquiring or resulting corporation and in which there is no material change to the interests held by the shareholders of the Company prior to such transaction and thereafter.
The Board may also determine that in the occurrence of a Fund-Raising Transaction (as defined below), that all of the outstanding and unexercised options held by or for the benefit of any grantee shall become fully vested. Such determination shall be specifically determined in the grantee’s letter of grant. “Fund-Raising Transaction” shall mean the raise by the Company of at least $10 million by way of public offerings and/or private placements of equity securities by one transaction or more, except in the event of issuance of equity securities in connection with the grant in exchange for services or as part of a commercial transaction.
114
In the event of termination of the employment or the director or service-provider relationship by us or by a related company within 12 months after a significant event in which the options were assumed, then the unvested portion of the options shall become fully vested and shall remain exercisable for a period of three months following the termination or notice of termination. For such purposes, a “Significant Event” would include our consolidation or merger with or into another corporation in which we are the ongoing or surviving corporation or in which, the ongoing or surviving corporation (or, if such transaction is effected through a subsidiary, the parent of such ongoing or surviving corporation) assumes the option or substitutes it with an appropriate option in the surviving corporation (or in the parent as aforesaid) in the manner set forth above.
| ITEM 7. | MAJOR SHAREHOLDERS AND RELATED PARTY TRANSACTIONS |
| A. | Major Shareholders |
The following table sets forth information with respect to the beneficial ownership of our ordinary shares as of March 15, 2019 by:
| ● | each of our directors and senior management; |
| ● | all of our directors and senior management as a group; and |
| ● | each person (or group of affiliated persons) known by us to be the beneficial owner of 5% or more of the outstanding ordinary shares. |
Beneficial ownership is determined in accordance with the rules of the SEC. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting or investment power with respect to those securities, and include shares subject to options and warrants that are exercisable within 60 days after March 15, 2019. Such shares are also deemed outstanding for purposes of computing the percentage ownership of the person holding the option, but not the percentage ownership of any other person.
Unless otherwise indicated below, to our knowledge, all persons named in the table have sole voting and investment power with respect to their shares, except to the extent that authority is shared by spouses under community property laws. None of our shareholders has informed us that he, she, or it is affiliated with a registered broker-dealer or is in the business of underwriting securities. None of our shareholders has different voting rights from other shareholders.
| Ordinary Shares Beneficially Owned |
Percentage Owned** |
|||||||
| Senior Management and Directors | ||||||||
| Jonathan M.N. Rigby (1) | - | - | ||||||
| Adi Goldin (2) | 539,806 | * | ||||||
| Abraham Havron (3) | 156,250 | * | ||||||
| Scott Burell (3) | 156,250 | * | ||||||
| Dr. Gili Hart (3) | 156,250 | * | ||||||
| Dr. Elan Penn (3) | 156,250 | * | ||||||
| Dr. Wolfgang Ruttenstorfer (4) | - | - | ||||||
| Yehiel Tal (5) | 4,500,639 | 2.3 | % | |||||
| Eran Rotem (6) | 1,832,827 | * | ||||||
| Oded Shoseyov (7) | 6,506,041 | 3.3 | % | |||||
| Philippe Bensimon (8) | 822,917 | * | ||||||
| Nadav Orr (9) | 680,208 | * | ||||||
| Ilana Belzer (10) | 438,542 | * | ||||||
| All senior management and directors as a group (13 persons) | 15,945,980 | 7.9 | % | |||||
| More than 5% Shareholders | ||||||||
| Meitav Dash Investment Ltd.(11) | 49,824,367 | 24.3 | % | |||||
| Docor Levi Lassen BV(12) | 10,609,639 | 5.5 | % | |||||
| Ami Sagy(13) | 46,377,017 | 23.2 | % | |||||
| Alpha Capital Anstalt(14) | 9,700,000 | 4.9 | % | |||||
| * | Less than 1% |
| ** | Based on 190,735,668 ordinary shares outstanding |
115
| (1) | Does not include 12,319,500 options to purchase 12,319,500 ordinary shares at an exercise price of $0.101 per share and expiring on January 30, 2026, the issuance of which is subject to shareholders’ approval. |
| (2) | Consists of: (i) 118,000 ordinary shares, (ii) 656,042 options to purchase 218,681 ordinary shares at an exercise price of NIS 1.80 per share and expiring on July 7, 2025, and (iii) 203,125 options to purchase 203,125 ordinary shares at an exercise price of NIS 0.58 per share and expiring on January 14, 2025. Does not include 250,000 options to purchase 250,000 ordinary shares at an exercise price of $0.101 per share and expiring on January 30, 2026, the issuance of which is subject to shareholders’ approval. |
| (3) | Consists of 156,250 options to purchase 156,250 ordinary shares at an exercise price of NIS 0.58 per share and expiring on January 14, 2025. Does not include 250,000 options to purchase 250,000 ordinary shares at an exercise price of $0.101 per share and expiring on January 30, 2026, the issuance of which is subject to shareholders’ approval. |
| (4) | Does not include 750,000 options to purchase 750,000 ordinary shares at an exercise price of $0.101 per share and expiring on January 30, 2026, the issuance of which is subject to shareholders’ approval. |
| (5) | Consists of (i) 1,505,875 ordinary shares, (ii) 153,041 options to purchase 51,014 ordinary shares exercisable at an exercise price of NIS 0.90 per share and expiring on May 3, 2020, (iii) 5,315,625 options to purchase 1,771,875 ordinary shares exercisable at an exercise price of NIS 1.80 per share and expiring on July 31, 2025, and (iv) 1,171,875 options to purchase 1,171,875 ordinary shares exercisable at an exercise price of NIS 0.58 per share and expiring on January 14, 2025. Does not include 2,700,000 options to purchase 2,700,000 ordinary shares at an exercise price of $0.101 per and expiring on January 30, 2026, the issuance of which is subject to shareholders’ approval. |
| (6) | Consists of (i) 450,000 options to purchase 150,000 ordinary shares exercisable at an exercise price of NIS 1.90 per share and expiring on October 20, 2021, (ii) 126,607 options to purchase 42,202 ordinary shares exercisable at an exercise price of NIS 0.90 per share and expiring on May 3, 2020, (iii) 2,812,500 options to purchase 937,500 ordinary shares exercisable at an exercise price of NIS 1.80 per share and expiring on May 15, 2025, and (iv) 703,125 options to purchase 703,125 ordinary shares exercisable at an exercise price of NIS 0.58 per share and expiring on December 26, 2024. Does not include 2,000,000 options to purchase 2,000,000 ordinary shares at an exercise price of $0.101 per share and expiring on January 30, 2026 that vest in more than 60 days of March 15, 2019. |
| (7) | Consists of (i) 2,737,573 ordinary shares, (ii) 2,258,813 options to purchase 752,938 ordinary shares at an exercise price of NIS 2.07 per share and expiring on February 16, 2021, (iii) 109,091 options to purchase 36,364 ordinary shares at an exercise price of NIS 0.90 per share and expiring on May 3, 2020, (iv) 8,000,000 options to purchase 2,666,667 ordinary shares at an exercise price of NIS 1.80 per share and expiring on July 31, 2025, and (v) 312,500 options to purchase 312,500 ordinary shares at an exercise price of NIS 0.58 per share and expiring on December 26, 2024. Does not include 1,000,000 options to purchase 1,000,000 ordinary shares at an exercise price of $0.101 per share and expiring on January 30, 2026 that vest in more than 60 days of March 15, 2019. |
116
| (8) | Consists of (i) 200,000 options to purchase 66,667 ordinary shares exercisable at an exercise price of NIS 4.17 per share and expiring on October 20, 2021, (ii) 300,000 options to purchase 100,000 ordinary shares exercisable at an exercise price of NIS 1.32 per share and expiring on May 3, 2020, (iii) 1,265,625 options to purchase 421,875 ordinary shares exercisable at an exercise price of NIS 1.80 per share and expiring on May 18, 2025, and (iv) 234,375 options to purchase 234,375 ordinary shares exercisable at an exercise price of NIS 0.58 per share and expiring on December 26, 2024. Does not include 800,000 options to purchase 800,000 ordinary shares at an exercise price of $0.101 per share and expiring on January 30, 2026 that vest in more than 60 days of March 15, 2019. |
| (9) | Consists of (i) 400,000 options to purchase 133,333 ordinary shares exercisable at an exercise price of NIS 0.76 per share and expiring on September 8, 2024, (ii) 937,500 options to purchase 312,500 ordinary shares exercisable at an exercise price of NIS 1.80 per share and expiring on May 18, 2025, and (iii) 234,375 options to purchase 234,375 ordinary shares exercisable at an exercise price of NIS 0.58 per share and expiring on December 26, 2024. Does not include 1,100,000 options to purchase 1,100,000 ordinary shares at an exercise price of $0.101 per share and expiring on January 30, 2026 that vest in more than 60 days of March 15, 2019. |
| (10) | Consists of (i) 612,500 options to purchase 204,167 ordinary shares exercisable at an exercise price of NIS 1.80 per share and expiring on August 31, 2025, and (ii) 234,375 options to purchase 234,375 ordinary shares exercisable at an exercise price of NIS 0.58 per share and expiring on December 26, 2024. Does not include 1,100,000 options to purchase 1,100,000 ordinary shares at an exercise price of $0.101 per share and expiring on January 30, 2026 that vest in more than 60 days of March 15, 2019. |
| (11) | Based partially on information contained in a Schedule 13G/A filed with the SEC on February 7, 2019 jointly by Meitav Dash Investments Ltd. and Meitav Dash Provident Funds and Pension Ltd. Consists of (i) 35,197,200 ordinary shares, (ii) 8,181,500 warrants to purchase 2,727,167 ordinary shares exercisable at an exercise price of NIS 1.20 per share and expiring on May 31, 2019, and (iii) a warrant to purchase 11,900,000 ordinary shares exercisable at an exercise price of NIS 0.80 per share and expiring on March 7, 2023. |
| (12) | To our knowledge, consists of (i) 9,847,639 ordinary shares, and (ii) 2,286,000 warrants to purchase 762,000 ordinary shares exercisable at an exercise price of NIS 1.20 per share and expiring on May 31, 2019. The ordinary shares are, to our knowledge, being held as follows: 5,543,305 by Docor Levi Lassen BV and 4,304,334 by its parent company, Docor International BV. To our knowledge, the Van Leer Foundation Group holds voting and dispositive control over the shares beneficially owned by Docor Levi Lassen BV and Docor International BV. |
| (13) | Based partially on information contained in a Schedule 13D filed with the SEC on February 11, 2019 by Ami Sagy. Consists of (i) 37,001,350 ordinary shares, (ii) 227,000 warrants to purchase 75,667 ordinary shares exercisable at an exercise price of NIS 1.2 per share and expiring on May 31, 2019, and (iii) a warrant to purchase 9,300,000 ordinary shares exercisable at an exercise price of NIS 0.80 per share and expiring on March 7, 2023. |
| (14) | To our knowledge, consists of (i) 6,173,840 ordinary shares, and (ii) 70,523 ADSs representing approximately 3,526,160 ordinary shares issuable upon exercise of prepaid warrants. Pursuant to the terms of certain warrants, the holder cannot convert such warrants if it would beneficially own, after any such conversion, more than 4.99% of the outstanding ordinary shares. The percentage in the table above gives effect to the blocker. Excludes (i) 770,561 ADSs representing approximately 38,528,063 ordinary shares issuable upon exercise of prepaid warrants within 60 days of March 15, 2019, and which are subject to the foregoing blocker, and (ii) 49,607,407 ordinary shares issuable upon exercise of a warrant. Konrad Ackerman has voting and dispositive power over the securities owned by Alpha. |
117
Bank of New York Mellon, or BNY, is the holder of record for our ADR program, pursuant to which each ADS represents 50 ordinary shares. As of March 15, 2019, BNY held 188,498,000 ordinary shares representing 99% of the outstanding ordinary shares at that date. Certain of these ordinary shares were held by brokers or other nominees. As a result, the number of holders of record or registered holders in the United States is not representative of the number of beneficial holders or of the residence of beneficial holders.
To our knowledge, from March 15, 2015 to March 15, 2019, the ownership percentage of Meitav Dash increased by 5.7% from 18.6% to 24.3%, the ownership percentage of Docor Levi Lassen BV decreased by 3.1% from 8.6% to 5.5% during such period, the ownership percentage of Ami Sagy increased by 16.4% from 6.8% to 23.2%. See “Item 7.B. Related Party Transactions” and “Item 10.C. Material Contracts” below for additional information.
| B. | Related Party Transactions |
The following is a description of the material terms of those transactions with related parties to which we are party and which were in effect since January 1, 2016.
U.S. dollar translations of NIS amounts are translated using the rate of NIS 3.748 to one U.S. dollar, the exchange rate reported by the Bank of Israel for December 31, 2018. All share amounts have been adjusted to give effect to the 1 for 3 reverse share split effected on November 20, 2016 while maintaining the exercise price of each option and warrant in effect prior to November 20, 2016, such that each option or warrant will be exercised for one third of one ordinary share of the Company. The descriptions provided below are summaries of the terms of such agreements and do not purport to be complete and are qualified in their entirety by the complete agreements.
We believe that we have executed all of our transactions with related parties on terms no less favorable to us than those we could have obtained from unaffiliated third parties. See “Item 6.C. Board Practices—Approval of Related Party Transactions under Israeli Law.”
Issuances of Securities
| ● | On June 9, 2016, in a financing we issued and sold 11,267,833 ordinary shares at a price per share of NIS 1.05 ($0.3), as well as 33,803,500 Series K warrants to purchase 11,267,833 ordinary shares at an exercise price of NIS 0.60 ($0.17) per warrant, for gross proceeds of NIS 11.8 million ($3.3 million). In addition, under the terms of the underwriting agreement, we issued 2,728,000 Series K warrants to purchase 909,333 ordinary shares to the Israeli underwriters in the transaction under the same conditions set out above. The following owners of our ordinary shares participated in these offerings: Docor Levi Lassen BV acquired 762,000 ordinary shares and 2,286,000 Series K warrants, and Meitav Dash acquired 2,727,167 ordinary shares and 8,181,500 Series K warrants. |
| ● | On February 12, 2017, we completed a public offering in which we sold 21,152,000 ordinary shares at a price per share of NIS 0.34, as well as 10,576,000 Series L warrants to purchase 10,576,000 ordinary shares at an exercise price of NIS 0.36 ($0.10) per warrant, for gross proceeds of NIS 7,191,680 ($2,037,880). The warrants were exercisable at NIS 0.36 per warrant until June 13, 2017. In addition, we issued 941,400 Series L warrants to purchase 941,400 ordinary shares to the underwriters in the transaction under the same conditions set out above. The following owners of our ordinary shares participated in these offerings: Meitav DS Investments Ltd, Docor International BV, Docor Levi Lassen BV, and Adi Goldin. During the second quarter of 2017, 10,055,464 Series L warrants were exercised into 10,055,464 ordinary shares at an exercise price of NIS 0.36 for each warrant resulting in NIS 3,618,000 ($1,025,220) in gross proceeds. 1,461,936 Series L warrants that were not exercised expired on June 14, 2017. |
| ● | On August 22, 2017, we issued to David Tsur, our former Chairman, 221,000 options to purchase 221,000 ordinary shares without an exercise price as well as an additional 265,000 options to purchase 265,000 ordinary shares with an exercise price of NIS 0.33 each. |
| ● | On September 6, 2017, we entered into the Alpha Purchase Agreement with Alpha, pursuant to which we agreed, upon the terms and subject to the conditions of the Alpha Purchase Agreement, to issue and sell to Alpha in a private placement, certain of our securities in three tranches, as follows: (i) at the first closing, ordinary shares and a Debenture, for a purchase price of $2,000,000, (ii) at the second closing, ordinary shares and/or a Debenture for a purchase price of $2,000,000 and (iii) at the third closing, ordinary shares and/or a Debenture, and a warrant to purchase 49,607,407 ordinary shares for a purchase price of $1,000,000. The first closing occurred on October 26, 2017, the second closing occurred on December 31, 2017, and the third closing occurred on April 30, 2018. For further information, see “Item 10. Additional Information—C. Material Contracts—Alpha Financing.” |
| ● | On November 8, 2017, we entered into the Meitav Purchase Agreement with Meitav Dash, pursuant to which we agreed, upon the terms and subject to the conditions of the Meitav Purchase Agreement, to issue and sell to Meitav Dash in a private placement, certain of our securities in three tranches, as follows: (i) at the first closing, 9,500,000 ordinary shares, for a purchase price of NIS 3,800,000 ($1,013,874), (ii) at the second closing, 2,400,000 ordinary shares for a purchase price of NIS 960,000 ($256,137), provided that Meitav Dash shall not be obligated to buy or hold, immediately following the second closing, 20% or more of our share capital and (iii) at the third closing for no additional consideration, warrants exercisable into 9,500,000 ordinary shares, and if the second closing has occurred, additional warrants exercisable into 2,400,000 ordinary shares. The first and second closings occurred on December 26, 2017 and the third closing occurred on March 7, 2018. For further information, see “Item 10. Additional Information—C. Material Contracts—Meitav Dash Financing.” |
| ● | On November 9, 2017, we entered into the Sagy Purchase Agreement with Ami Sagy pursuant to which we agreed, upon the terms and subject to the conditions of the Sagy Purchase Agreement, to issue and sell to Ami Sagy in a private placement, certain of our securities in two tranches, as follows: (i) at the first closing, 9,300,000 ordinary shares, for a purchase price of NIS 3,720,000 ($992,529) and (ii) at the second closing for no additional consideration, warrants exercisable into 9,300,000 ordinary shares, or the Sagy Warrants. The first closing occurred on December 26, 2017 and the second closing occurred on March 7, 2018. For further information, see “Item 10. Additional Information—C. Material Contracts—Ami Sagy Financing.” |
118
| ● | On December 7, 2017, our board of directors approved the grant of 16,050,000 options to purchase 16,050,000 ordinary shares at an exercise price of NIS 0.58 ($0.15) per option to certain officers, directors and employees. On January 14, 2018, our shareholders approved the grant to the directors and to our CEO. |
| ● | On January 18, 2018, we entered into Security Purchase Agreements for the purchase and sale, in a private placement, of an aggregate of 4,344,340 ordinary shares for an aggregate of NIS 2,172,170 ($579,554) to the following three investors as follows: (i) Alpha entered into a Security Purchase Agreement for the purchase and sale of 1,275,340 ordinary shares for NIS 637,670 ($170,136); (ii) Ami Sagy entered into a Security Purchase Agreement for the purchase and sale of 2,046,000 ordinary shares for NIS 1,023,000 ($272,946); and (iii) Docor International BV entered into a Security Purchase Agreement for the purchase and sale of 1,023,000 ordinary shares for NIS 511,500 ($136,473). Closing occurred on January 25, 2018. | |
| ● | On July 26, 2018, we entered into a Securities Purchase Agreement with Ami Sagy for the purchase and sale, in a private placement, of 11,125,000 ordinary shares for an aggregate purchase price of NIS 4,561,250 (approximately $1.2 million). Closing occurred on July 31, 2018. |
Agreements with Yissum
We have entered into certain agreements with Yissum, in which Prof. Oded Shoseyov, our Chief Scientist, who served as our Chief Scientific Officer until March 26, 2019, has or might have a personal interest, including an agreement dated July 13, 2004 with respect to the intellectual property rights relating to our rhCollagen. See “Item 4.B. Business Overview— Intellectual Property—Agreement with Yissum Research Development Company of the Hebrew University of Jerusalem Ltd. with Respect to Our rhCollagen,” and see “Item 6.C. Board Practices—Approval of Related Party Transactions Under Israeli Law.”
On July 29, 2010, we signed a joint development and cross license agreement with Yissum, which agreement was amended on September 4, 2017. The agreement governs the relationship between the parties in connection with the invention protected by a patent application for the Resilin protein and future results from development work related to Resilin conducted jointly by us and Yissum or solely by us or Yissum. The Resilin protein and its patent are not related to our collagen protein and its related patents. The agreement stipulates that the parties will be co-owners of the Resilin patent and its associated know-how developed prior to the date of execution of the agreement. Developments results developed by the company together with Yissum, or independently by Yissum within the company’s field shall be jointly owned by both parties. Developments results developed independently by the company, or independently by Yissum in Yissum’s field, shall be owned by the developing party. Each party has granted the other an exclusive worldwide license, which can be sub-licensed, to make use of the Resilin patent and its associated know-how, including the joint IP developed under this agreement, for the purposes of research, development, production, marketing, distribution, license or sale of products limited to the licensee’s field of use. Accordingly, per the agreement as amended, we have exclusive rights to the technology for all medical and cosmetic human uses (including, without limitation therapeutic, aesthetic, skin care and diagnostic uses but not including hair straightening and nail coating uses) and veterinary uses. Yissum has exclusivity in any other field. We were also granted first rights to develop and commercialize products in Yissum’s field of exclusivity where a sub-license has not yet been given by Yissum to a third party.
On April 20, 2015, we entered into a consortium agreement with several international companies and academic institutions, outlining the framework of a tissue research and development project using nanotechnology, our rhCollagen, and stem cell technology. The project is expected to last approximately three years. The Hebrew University of Jerusalem together with Yissum and Prof. Oded Shoseyov and the project manager on behalf of Yissum, will also take part in the project.
As part of the project, we will supply an insignificant amount of our rhCollagen to the Hebrew University, and become a member of the steering committee of the project. The agreement contains provisions protecting each consortium member’s rights including with respect to the intellectual property to be developed as part of the project, and protecting us, our rhCollagen, and any intellectual property developed as part of the project with respect to our rhCollagen whether by the Hebrew University or by other parties participating in the consortium, as applicable.
119
Rights of Appointment
Our current board of directors currently consists of seven directors. See “Item 6.A.—Directors and Senior Management.” Currently serving directors that were appointed (other than the external directors) will continue to serve pursuant to their appointment until the next annual meeting of shareholders.
The Alpha Purchase Agreement provided for certain board appointment rights. On the first closing, we were required to appoint two directors selected by Alpha (out of a seven-member board), and on the second closing, we were required to appoint one director selected by Alpha (out of an eight-member board). At the first closing, Alpha selected Scott Burell to serve on the board, and in June 2018, Alpha selected Dr. Wolfgang Ruttenstorfer to serve on the board.
Registration Rights
In connection with the first closing of the Alpha financing, we entered into a Registration Rights Agreement with Alpha. Pursuant to the Registration Rights Agreement, we agreed to file a registration statement with the SEC within 45 days from the date of the Registration Rights Agreement to register the resale of our ordinary shares held by Alpha that were issued in the private placement including ordinary shares underlying the Debentures, Warrants and pre-paid warrants and to maintain the effectiveness thereunder. We also agreed to use best efforts to have the registration statement declared effective within 105 days from the date of the Registration Rights Agreement and use best efforts to keep the registration statement continuously effective until the earlier of (i) the date after which all of the securities to be registered thereunder have been sold, or (ii) the date on which all the securities to be registered thereunder may be sold without volume or manner-of-sale restrictions and without current public information pursuant to Rule 144 under the Securities Act.
Agreements with Directors and Senior Management
Insurance, Exculpation, and Indemnification Agreements
We have entered into indemnification agreements with each of our current directors and executive officers exculpating them from a breach of their duty of care to us to the fullest extent permitted by law, subject to limited exceptions, and undertaking to indemnify them to the fullest extent permitted by Israeli law, subject to limited exceptions, and including with respect to liabilities resulting from this offering to the extent such liabilities are not covered by insurance. See “Item 6.C. Board Practices—Approval of Related Party Transactions Under Israeli Law—Exculpation, Insurance and Indemnification of Directors and Officers.”
Employment and Services Agreements
We have entered into employment or services agreements with our senior management. See “Item 6.B. Compensation.”
Options
We have granted options to purchase our ordinary shares to certain of our officers and directors. See “Item 6.B. Compensation” and “Item 7.A. Major Shareholders.” We describe our option plans under “Item 6.E. Share Ownership” and “Item 7.A. Major Shareholders.”
| C. | Interests of Experts and Counsel |
Not applicable.
| ITEM 8. | FINANCIAL INFORMATION. |
| A. | Consolidated Statements and Other Financial Information. |
See “Item 18. Financial Statements.”
120
Legal Proceedings
See “Item 4.B. Business Overview―Legal Proceedings.”
Dividends
We have never declared or paid cash dividends to our shareholders. Currently, we do not intend to pay cash dividends. We intend to reinvest any earnings in developing and expanding our business. Any future determination relating to our dividend policy will be at the discretion of our board of directors and will depend on a number of factors, including future earnings, our financial condition, operating results, contractual restrictions, capital requirements, business prospects, applicable Israeli law and other factors our board of directors may deem relevant. In addition, the distribution of dividends is limited by Israeli law, which permits the distribution of dividends only out of distributable profits.
If we pay any dividends, we will also pay such dividends to the ADS holders to the same extent as holders of our ordinary shares, subject to the terms of the deposit agreement, including the fees and expenses payable thereunder. No dividends will accrue for any unexercised warrants. Cash dividends on our ordinary shares, if any, will be paid to ADS holders in U.S. dollars.
| B. | Significant Changes |
Other than as otherwise described in this Annual Report on Form 20-F and as set forth below, no significant change has occurred in our operations since the date of our consolidated financial statements included in this Annual Report on Form 20-F.
| ITEM 9. | THE OFFER AND LISTING |
| A. | Offer and Listing Details |
On January 31, 2018, our ADSs commenced trading on the Nasdaq Capital Market under the symbol “CLGN.” Our ADSs were quoted on the OTCQX from March 2015 to May 25, 2017, and quoted on the OTCQB from May 26, 2017 to January 30, 2018.
| B. | Plan of Distribution |
Not applicable.
| C. | Markets |
Our ADSs are listed on the Nasdaq Capital Market.
| D. | Selling Shareholders |
Not applicable.
| E. | Dilution |
Not applicable.
| F. | Expenses of the Issue |
Not applicable.
121
| ITEM 10. | ADDITIONAL INFORMATION |
| A. | Share Capital |
Not applicable.
| B. | Memorandum and Articles of Association |
Articles of Association
The following are summaries of material provisions of our articles of association and the Companies Law insofar as they relate to the material terms of our ordinary shares.
Purposes and Objects of the Company
Our purpose as set forth in our articles of association is to engage in any lawful activity.
Registration Number
Our registration number with the Israeli Registrar of Companies is 52-0039785.
Voting Rights and Conversion
All ordinary shares have identical voting and other rights in all respects.
Transfer of Shares
Our fully paid ordinary shares are issued in registered form and may be freely transferred under our articles of association, unless the transfer is restricted or prohibited by another instrument, applicable law, or the rules of a stock exchange on which the shares are listed for trade. The ownership or voting of our ordinary shares by non-residents of Israel is not restricted in any way by our articles of association or the laws of the State of Israel, except for ownership by nationals of some countries that are, or have been, in a state of war with Israel.
Election of Directors
Our ordinary shares do not have cumulative voting rights for the election of directors. As a result, the holders of a majority of the voting power represented at a shareholders meeting have the power to elect all of our directors, subject to the special approval requirements for external directors described under “Management—External Directors.”
Under our articles of association, our board of directors must consist of not less than three but no more than twelve directors, including two external directors, as required by the Companies Law. Pursuant to our articles of association, other than the external directors, for whom special election requirements apply under the Companies Law, the vote required to appoint a director is a simple majority vote of holders of our voting shares, participating and voting at the relevant meeting. Each director will serve until his or her successor is duly elected and qualified or until his or her earlier death, resignation, or removal by a vote of the majority voting power of our shareholders at a general meeting of our shareholders or until his or her office expires by operation of law, in accordance with the Companies Law. In addition, our articles of association allow our board of directors to appoint directors to fill vacancies on the board of directors to serve for a term of office equal to the remaining period of the term of office of the directors(s) whose office(s) have been vacated. External directors are elected for an initial term of three years, may be elected for additional terms of three years each under certain circumstances, and may be removed from office pursuant to the terms of the Companies Law. Our board of directors has decided to adopt an exemption that provides relief for Israeli companies whose shares are listed on certain stock exchanges outside of Israel (including the Nasdaq Capital Market) with no controlling shareholder, such as ourselves, from being required to appoint external directors so long as such companies satisfy the requirements of the foreign laws in the listing jurisdiction outside of Israel which apply to companies incorporated in such jurisdiction, in respect of the appointment of independent directors and the composition of the audit committee and compensation committee. An amendment to our articles of association that reflects such exemption is subject to shareholders approval, which is pending. See “Item 6.A. Directors and Senior Management—External Directors.” for more information.
122
Dividend and Liquidation Rights
We may declare a dividend to be paid to the holders of our ordinary shares in proportion to their respective shareholdings. Under the Companies Law, dividend distributions are determined by the board of directors and do not require the approval of the shareholders of a company unless the company’s articles of association provide otherwise. Our articles of association do not require shareholder approval of a dividend distribution and provide that dividend distributions may be determined by our board of directors.
Pursuant to the Companies Law, the distribution amount is limited to the greater of retained earnings or earnings generated over the two most recent fiscal years, according to our then last reviewed or audited financial statements, provided that the date of the financial statements is not more than six months prior to the date of the distribution, or we may otherwise only distribute dividends that do not meet such criteria only with court approval. In each case, we are only permitted to distribute a dividend if our board of directors or the court, if applicable, determines that there is no reasonable concern that payment of the dividend will prevent us from satisfying our existing and foreseeable obligations as they become due.
In the event of our liquidation, after satisfaction of liabilities to creditors, our assets will be distributed to the holders of our ordinary shares in proportion to their shareholdings. This right, as well as the right to receive dividends, may be affected by the grant of preferential dividend or distribution rights to the holders of a class of shares with preferential rights that may be authorized in the future.
With respect to non-exculpation of a director from liability arising out of a prohibited dividend or distribution to shareholders see “Item 6.C. Board Practices—Approval of Related Party Transactions Under Israeli Law—Exculpation, Insurance and Indemnification of Directors and Officers.”
Shareholder Meetings
Under Israeli law, we are required to hold an annual general meeting of our shareholders once every calendar year that must be held no later than 15 months after the date of the previous annual general meeting. All meetings other than the annual general meeting of shareholders are referred to in our articles of association as extraordinary general meetings. Our board of directors may call extraordinary general meetings whenever it sees fit, at such time and place, within or outside of Israel, as it may determine. In addition, the Companies Law provides that our board of directors is required to convene an extraordinary general meeting upon the written request of (i) any two of our directors or one-fifth of the members of our board of directors or (ii) one or more shareholders holding, in the aggregate, either (a) 5% or more of our outstanding issued shares and 1% of our outstanding voting power or (b) 5% or more of our outstanding voting power. One or more shareholders, holding 1% or more of the outstanding voting power, may ask the board to add an item to the agenda of a prospective meeting, if the proposal merits discussion at the general meeting.
Subject to the provisions of the Companies Law and the regulations promulgated thereunder, shareholders entitled to participate and vote at general meetings are the shareholders of record on a date to be decided by the board of directors, which may be between four and 40 days prior to the date of the meeting. Furthermore, the Companies Law requires that resolutions regarding the following matters must be passed at a general meeting of our shareholders:
| ● | amendments to our articles of association; |
| ● | appointment or termination of our auditors; |
| ● | appointment of external directors; |
| ● | approval of certain related party transactions; |
| ● | increases or reductions of our authorized share capital; |
| ● | a merger; and |
| ● | the exercise of our board of director’s powers by a general meeting, if our board of directors is unable to exercise its powers and the exercise of any of its powers is required for our proper management. |
123
The Companies Law and the regulations thereof require that a notice of any annual general meeting or extraordinary general meeting be provided to shareholders at least 21 days or 14 days, as applicable, prior to the meeting and if the agenda of the meeting includes, for example, the appointment or removal of directors, the approval of transactions with office holders or interested or related parties, or an approval of a merger, notice must be provided at least 35 days prior to the meeting.
All shareholder decisions are to be taken by votes in a shareholders’ meeting. Under the Companies Law and our articles of association, shareholders are not permitted to take action via written consent in lieu of a meeting.
Voting Rights
Quorum Requirements
Pursuant to our articles of association, holders of our ordinary shares have one vote for each ordinary share held on all matters submitted to a vote before the shareholders at a general meeting. As a foreign private issuer, the quorum required for our general meetings of shareholders consists of at least two shareholders present in person, by proxy, or written ballot who hold or represent between them at least 20% of the total outstanding voting rights. A meeting adjourned for lack of a quorum is generally adjourned to the same day in the following week at the same time and place or to a later time or date if so specified in the notice of the meeting. At the reconvened meeting, any two or more shareholders present in person or by proxy shall constitute a lawful quorum. See “Item 16G.—Corporate Governance Practices” for more information.
Vote Requirements
Our articles of association provide that all resolutions of our shareholders require a simple majority vote, unless otherwise required by the Companies Law or by our articles of association. Under the Companies Law, each of (i) the approval of an extraordinary transaction with a controlling shareholder and (ii) the terms of employment or other engagement of the controlling shareholder of the company or such controlling shareholder’s relative (even if not extraordinary) requires the approval described above under “Management—Approval of Related Party Transactions Under Israeli Law—Disclosure of Personal Interests of Controlling Shareholders and Approval of Certain Transactions.” Under our articles of association, the alteration of the rights, privileges, preferences, or obligations of any class of our shares requires a simple majority vote of the class so affected (or such other percentage of the relevant class that may be set forth in the governing documents relevant to such class), in addition to the ordinary majority vote of all classes of shares voting together as a single class at a shareholder meeting. An exception to the simple majority vote requirement is a resolution for the voluntary winding up, or an approval of a scheme of arrangement or reorganization, of the company pursuant to Section 350 of the Companies Law, which requires the approval of holders of 75% of the voting rights represented at the meeting, in person, by proxy, or by voting deed and voting on the resolution.
Access to Corporate Records
Under the Companies Law, shareholders are provided access to: minutes of our general meetings; our shareholders register and principal shareholders register, articles of association and financial statements; and any document that we are required by law to file publicly with the Israeli Companies Registrar or the ISA. In addition, shareholders may request to be provided with any document related to an action or transaction requiring shareholder approval under the related party transaction provisions of the Companies Law. We may deny this request if we believe it has not been made in good faith or if such denial is necessary to protect our interest or protect a trade secret or patent.
Modification of Class Rights
Under the Companies Law and our articles of association, the rights attached to any class of share, such as voting, liquidation, and dividend rights, may be amended by adoption of a resolution by the holders of a majority of the shares of that class present at a separate class meeting, or otherwise in accordance with the rights attached to such class of shares, as set forth in our articles of association.
124
Registration Rights
In connection with the first closing of the Alpha financing, we entered into a Registration Rights Agreement with Alpha. Pursuant to the Registration Rights Agreement, we agreed to file a registration statement with the SEC within 45 days from the date of the Registration Rights Agreement to register the resale of our ordinary shares held by Alpha that were issued in the private placement including ordinary shares underlying the Debentures, Warrants and pre-paid warrants and to maintain the effectiveness thereunder. We also agreed to use best efforts to have the registration statement declared effective within 105 days from the date of the Registration Rights Agreement and use best efforts to keep the registration statement continuously effective until the earlier of (i) the date after which all of the securities to be registered thereunder have been sold, or (ii) the date on which all the securities to be registered thereunder may be sold without volume or manner-of-sale restrictions and without current public information pursuant to Rule 144 under the Securities Act.
Acquisitions under Israeli Law
Full Tender Offer
A person wishing to acquire shares of an Israeli public company and who would as a result hold over 90% of the target company’s issued and outstanding share capital is required by the Companies Law to make a tender offer to all of the company’s shareholders for the purchase of all of the issued and outstanding shares of the company. A person wishing to acquire shares of a public Israeli company and who would, as a result, hold over 90% of the issued and outstanding share capital of a certain class of shares is required to make a tender offer to all of the shareholders who hold shares of the relevant class for the purchase of all of the issued and outstanding shares of that class. If the shareholders who do not accept the offer hold less than 5% of the issued and outstanding share capital of the company or of the applicable class, and more than half of the shareholders who do not have a personal interest in the offer accept the offer, all of the shares that the acquirer offered to purchase will be transferred to the acquirer by operation of law. However, a tender offer will also be accepted if the shareholders who do not accept the offer hold less than 2% of the issued and outstanding share capital of the company or of the applicable class of shares.
Upon a successful completion of such a full tender offer, any shareholder that was an offeree in such tender offer, whether such shareholder accepted the tender offer or not, may, within six months from the date of acceptance of the tender offer, petition an Israeli court to determine whether the tender offer was for less than fair value and that the fair value should be paid as determined by the court. However, under certain conditions, the offeror may include in the terms of the tender offer that an offeree who accepted the offer will not be entitled to petition the Israeli court as described above.
If (i) the shareholders who did not respond or accept the tender offer hold at least 5% of the issued and outstanding share capital of the company or of the applicable class or the shareholders who accept the offer constitute less than a majority of the offerees that do not have a personal interest in the acceptance of the tender offer, or (ii) the shareholders who did not accept the tender offer hold 2% or more of the issued and outstanding share capital of the company (or of the applicable class), the acquirer may not acquire shares of the company that will increase its holdings to more than 90% of the company’s issued and outstanding share capital or of the applicable class from shareholders who accepted the tender offer.
Special Tender Offer
The Companies Law provides that an acquisition of shares of an Israeli public company must be made by means of a special tender offer if as a result of the acquisition the purchaser would become a holder of 25% or more of the voting rights in the company. This requirement does not apply if there is already another holder of at least 25% of the voting rights in the company. Similarly, the Companies Law provides that an acquisition of shares in a public company must be made by means of a special tender offer if, as a result of the acquisition, the purchaser would become a holder of more than 45% of the voting rights in the company, provided that there is no other shareholder of the company who holds more than 45% of the voting rights in the company, subject to certain exceptions.
125
A special tender offer must be extended to all shareholders of a company but the offeror is not required to purchase shares representing more than 5% of the voting power attached to the company’s outstanding shares, regardless of how many shares are tendered by shareholders. A special tender offer may be consummated only if (i) outstanding shares representing at least 5% of the voting power of the company will be acquired by the offeror and (ii) the number of shares tendered in the offer exceeds the number of shares whose holders objected to the offer (excluding the purchaser, controlling shareholders, holders of 25% or more of the voting rights in the company or any person having a personal interest in the acceptance of the tender offer). If a special tender offer is accepted, then the purchaser or any person or entity controlling it or under common control with the purchaser or such controlling person or entity may not make a subsequent tender offer for the purchase of shares of the target company and may not enter into a merger with the target company for a period of one year from the date of the offer, unless the purchaser or such person or entity undertook to effect such an offer or merger in the initial special tender offer.
Under the Companies Regulations (Relief for Public Companies whose Shared are Traded on Exchanges outside of Israel), the above requirements for a special tender offer do not apply in instances whereby according to the laws of the foreign jurisdiction there are limitations regarding the acquisition of a controlling interest in the company of any specified portion or the acquisition of a controlling interest of any specified portion necessitates an offer by the potential acquirer of a controlling interest to acquire shares from amongst the publicly traded shares.
Merger
The Companies Law permits merger transactions if approved by each party’s board of directors and, unless certain requirements described under the Companies Law are met, by a majority vote of each party’s shareholders, and, in the case of the target company, a majority vote of each class of its shares voted on the proposed merger at a shareholders meeting.
The board of directors of a merging company is required pursuant to the Companies Law to discuss and determine whether, in its opinion, there exists a reasonable concern that, as a result of a proposed merger, the surviving company will not be able to satisfy its obligations towards its creditors, taking into account the financial condition of the merging companies. If the board of directors has determined that such a concern exists, it may not approve a proposed merger. Following the approval of the board of directors of each of the merging companies, the boards of directors must jointly prepare a merger proposal for submission to the Israeli Registrar of Companies.
For purposes of the shareholder vote, unless a court rules otherwise, the merger will not be deemed approved if a majority of the votes of shares represented at the shareholders meeting that are held by parties other than the other party to the merger, or by any person (or group of persons acting in concert) who holds (or hold, as the case may be) 25% or more of the voting rights or the right to appoint 25% or more of the directors of the other party, vote against the merger. If, however, the merger involves a merger with a company’s own controlling shareholder or if the controlling shareholder has a personal interest in the merger, then the merger is instead subject to the same special majority approval that governs all extraordinary transactions with controlling shareholders (as described under “Management—Approval of Related Party Transactions Under Israeli Law—Disclosure of Personal Interests of Controlling Shareholders and Approval of Certain Transactions”).
If the transaction would have been approved by the shareholders of a merging company but for the separate approval of each class or the exclusion of the votes of certain shareholders as provided above, a court may still approve the merger upon the request of holders of at least 25% of the voting rights of a company, if the court holds that the merger is fair and reasonable, taking into account the value of the parties to the merger and the consideration offered to the shareholders of the target company.
Upon the request of a creditor of either party to the proposed merger, the court may delay or prevent the merger if it concludes that there exists a reasonable concern that, as a result of the merger, the surviving company will be unable to satisfy the obligations of the merging entities, and may further give instructions to secure the rights of creditors.
126
In addition, a merger may not be consummated unless at least 50 days have passed from the date on which a proposal for approval of the merger was filed by each party with the Israeli Registrar of Companies and at least 30 days have passed from the date on which the merger was approved by the shareholders of each party.
Borrowing Powers
Pursuant to the Companies Law and our articles of association, our board of directors may exercise all powers and take all actions that are not required under law or under our articles of association to be exercised or taken by our shareholders, including the power to borrow money for company purposes.
Changes in Capital
Our articles of association enable us to increase or reduce our share capital. Any such changes are subject to the provisions of the Companies Law and must be approved by a resolution duly passed by our shareholders at a general meeting by voting on such change in the capital. In addition, certain transactions that have the effect of reducing capital, such as the declaration and payment of dividends in the absence of sufficient retained earnings or profits, require the approval of both our board of directors and an Israeli court.
| C. | Material Contracts |
Except as set forth below, we have not entered into any material contract within the two years prior to the date of this Annual Report on Form 20-F, other than contracts entered into in the ordinary course of business, or as otherwise described herein in “Item 4.A. History and Development of the Company”, “Item 4.B. Business Overview”, “Item 7A. Major Shareholders” or “Item 7B. Related Party Transactions” above.
February 2017 Financing
On February 12, 2017, we completed a public offering in which we sold 21,152,000 ordinary shares at a price per share of NIS 0.34, as well as 10,576,000 Series L warrants to purchase 10,576,000 ordinary shares at an exercise price of NIS 0.36 ($0.10) per warrant, for gross proceeds of NIS 7,191,680 ($1,918,804). The warrants were exercisable at NIS 0.36 per warrant until June 13, 2017. In addition, we issued 941,400 Series L warrants to purchase 941,400 ordinary shares to the underwriters in the transaction under the same conditions set out above. The following owners of our ordinary shares participated in these offerings: Meitav Investments Ltd, Docor International BV, Docor Levi Lassen BV, and Adi Goldin, the Chairman of the Company’s board of directors.
During the second quarter of 2017, 10,055,464 Series L warrants were exercised into 10,055,464 ordinary shares at an exercise price of NIS 0.36 for each warrant resulting in NIS 3,618,000 ($965,315) in gross proceeds. 1,461,936 Series L warrants that were not exercised expired on June 14, 2017.
Alpha Financing
On September 6, 2017, we entered into the Alpha Purchase Agreement with Alpha, pursuant to which we agreed, upon the terms and subject to the conditions of the Alpha Purchase Agreement, to issue and sell to Alpha in a private placement, certain of our securities in three tranches, as follows: (i) at the first closing, ordinary shares and a Convertible Debenture, or Debenture, for a purchase price of $2,000,000, (ii) at the second closing, ordinary shares and/or a Debenture for a purchase price of $2,000,000, and (iii) at the third closing, ordinary shares and/or a Debenture, and the Alpha Warrant, for a purchase price of $1,000,000.
Alpha Purchase Agreement
At each closing, the number of ordinary shares issuable was calculated by dividing the applicable purchase price by NIS 0.36144, subject to adjustment for share splits, share dividends, and the like, and with respect to each of the first and second closings, an additional 3,458,408 ordinary shares are issuable for no cash consideration; provided that to the extent that the purchaser’s ownership of ordinary shares, together with any of its affiliates, would exceed a beneficial ownership limitation of 4.99%, then Alpha may, at its option, elect to apply the applicable purchase price to the purchase of Debentures.
127
We completed the first closing on October 26, 2017, which resulted in the issuance to Alpha of an aggregate of 7,280,000 ordinary shares and a Debenture in the principal amount of $1,375,144 for gross proceeds of $2,000,000. We completed the second closing on December 31, 2017, which resulted in the issuance to Alpha of a Debenture in the principal amount of $2,000,000. Upon the listing of our ADSs on the Nasdaq Capital Market, the Debentures automatically converted into a pre-paid warrant to purchase 786,455 ADSs representing 39,322,742 ordinary shares. During July and August 2018, Alpha exercised a portion of such pre-paid warrant into 165,000 ADSs representing 8,250,000 ordinary shares. We completed the third closing on April 30, 2018, which resulted in the issuance to Alpha of a pre-paid warrant to purchase 9,921,482 ordinary shares represented by 198,430 ADSs and the Alpha Warrant to purchase up to 49,607,407 ordinary shares represented by 992,149 ADSs, at an exercise price of NIS 36.14 per ADS ($10.28 per ADS), for gross proceeds of $1 million.
Under the Alpha Purchase Agreement, Alpha was granted a right of participation in certain future offerings until October 26, 2018. In addition, the Alpha Purchase Agreement contains full-ratchet anti-dilution protection until October 26, 2019 in the event of certain subsequent equity issuances at a price that is lower than the then applicable per ordinary share purchase price.
The Alpha Purchase Agreement provides for the following restrictions on future issuances of securities (subject to certain exempt issuances): (i) until the 24 month anniversary of the second closing or the applicable date of termination of the Alpha Purchaser Agreement pursuant to the terms therein, if applicable, (as the case may be), we are prohibited from effecting a variable rate transaction, (ii) until the 12 month anniversary of the third closing, we are prohibited from issuing any equity securities that include any anti-dilution protection (other than customary anti-dilution protection for share splits, dividends and the like), and (iii) until the 12 month anniversary of the second closing or the applicable date of termination of the Alpha Purchaser Agreement pursuant to the terms therein, if applicable, (as the case may be), we are prohibited from issuing any equity securities for an effective price per share less than the effective per ordinary purchase price, subject to adjustment for share splits, dividends and the like.
The Alpha Purchase Agreement further provides for certain board appointment rights. On the first closing, we were required to appoint two directors selected by Alpha (out of a seven-member board) and on the second closing, we were required to appoint one director selected by Alpha (out of an eight-member board), each who shall serve as directors at least until the end of our 2018 annual general meeting. At the first closing, Alpha selected Scott Burell to serve on the board, and in June 2018, Alpha selected Dr. Wolfgang Ruttenstorfer to serve on the board.
We were required under the Alpha Purchase Agreement to use commercially reasonable efforts to take the necessary steps to transition to dual-listing reporting format with a view to delisting our ordinary shares from the TASE and to list the ADSs on the Nasdaq Capital Market. We delisted our ordinary shares from the TASE, and the last date of trading of our ordinary shares was on October 29, 2018.
If we fail to timely effect a legend removal in accordance with the Alpha Purchase Agreement, the Alpha Purchase Agreement provides for certain liquidated damages and customary buy-in provisions. In addition, the Alpha Purchase Agreement provides for certain liquidated damages in the case of a failure to satisfy certain current public information requirements under Rule 144.
The Alpha Purchase Agreement also contains representations and warranties, covenants and indemnification provisions customary in transactions of this nature.
Debentures
In connection with the first and second closings, we issued Debentures in an aggregate principal amount of $3,375,144. As stated above, upon the listing of our ADSs on the Nasdaq Capital Market, the Debentures automatically converted into a pre-paid warrant to purchase 786,455 ADSs representing approximately 39,322,742 ordinary shares.
128
The Debenture issuable had a maturity date of five years from the date of issuance and is interest-free. The Debenture was convertible at any time at the option of the holder into ADSs at a conversion price of the US dollar equivalent, as for the Debenture issued in the first and second closing, of NIS 15.3897 and, for the Debenture issued in the third closing, of NIS 18.0719 (each calculated in accordance with the rate of exchange of NIS 3.586 per US$1.00) per ADS. In addition, the Debenture was mandatorily convertible at the then effective conversion price without regard to any beneficial ownership limitation if (i) the ADSs or our ordinary shares are approved for listing on the Nasdaq Capital Market, and (ii) certain equity conditions are met, including, among other things, an effective registration covering a minimum number of ordinary shares held by the holder or that all the ordinary shares or ADSs held by the holder may be sold under Rule 144 without volume or manner-of-sale restrictions or current public information requirements; provided that the holder could elect to convert the Debenture in whole or in part to a pre-paid warrant to purchase such number of ADSs otherwise issuable upon mandatory conversion of the Debenture. The pre-paid warrant may be exercised on a cashless basis at any time. The pre-paid warrant is subject to certain anti-dilution adjustments upon certain events, including share splits, share dividends, subsequent rights offerings, pro-rata distributions and fundamental transactions. In addition, we entered into a side letter with Alpha pursuant to which any ordinary shares or ADSs issued upon exercise of a pre-paid warrant are subject to full-ratchet anti-dilution protection until October 26, 2019 in the event of certain subsequent equity issuances at a price that is lower than the applicable conversion price of the Debenture.
The Debenture was subject to certain anti-dilution adjustments upon certain events, including share splits, share dividends, subsequent rights offerings, pro-rata distributions and fundamental transactions. In addition, the Debenture contained full-ratchet anti-dilution protection until October 26, 2019 in the event of certain subsequent equity issuances at a price that is lower than the then applicable conversion price.
Upon the occurrence of certain events of default, the outstanding principal amount of the Debenture, together with other amounts due, would become, at the election of the holder, immediately due and payable in cash at the “Mandatory Default Amount” as defined in the Debenture. In addition, if we fail to timely effectuate a conversion under the terms of the Debenture, the Debenture provided for certain liquidated damages and customary buy-in provisions.
The Debenture was an unsecured, general obligation and ranks pari passu with other unsecured and unsubordinated liabilities. As stated above, upon the listing of our ADSs on the Nasdaq Capital Market, the Debentures automatically converted into pre-paid warrants to purchase 786,455 ADSs representing approximately 39,322,742 ordinary shares.
Warrant
At the third closing, we issued the Alpha Warrant to purchase 49,607,407 ordinary shares represented by 992,149 ADSs. The Alpha Warrant may be exercised for a period of five years from issuance at an exercise price of the US dollar equivalent of NIS 36.14379 per ADS (calculated in accordance with the known representative rate of exchange on the date of the notice of exercise). The Alpha Warrant may be exercised on a cashless basis if after the one-year anniversary of issuance there is no effective registration statement covering the resale of the ADSs underlying the Alpha Warrant.
The Alpha Warrant is subject to certain anti-dilution adjustments upon certain events, including share splits, share dividends, subsequent rights offerings, pro-rata distributions and fundamental transactions (which, in the case of fundamental transactions, is subject to certain limitations). In addition, the Alpha Warrant contains full-ratchet anti-dilution protection until October 26, 2019 in the event of certain subsequent equity issuances at a price that is lower than the applicable exercise price of the Alpha Warrant.
If we fail to timely effectuate an exercise under the terms of the Alpha Warrant, the Alpha Warrant provides for certain liquidated damages and customary buy-in provisions.
Registration Rights Agreement
In connection with the first closing of the Alpha financing, we entered into a Registration Rights Agreement with Alpha. Pursuant to the Registration Rights Agreement, we agreed to file a registration statement with the SEC within 45 days from the date of the Registration Rights Agreement to register the resale of our ordinary shares held by Alpha that were issued in the private placement including ordinary shares underlying the Debentures, Warrants and pre-paid warrants and to maintain the effectiveness thereunder. We also agreed to use best efforts to have the registration statement declared effective within 105 days from the date of the Registration Rights Agreement and use best efforts to keep the registration statement continuously effective until the earlier of (i) the date after which all of the securities to be registered thereunder have been sold, or (ii) the date on which all the securities to be registered thereunder may be sold without volume or manner-of-sale restrictions and without current public information pursuant to Rule 144 under the Securities Act.
129
Meitav Dash Financing
On November 8, 2017, we entered into a securities purchase agreement, or the Meitav Purchase Agreement, with Meitav Dash, pursuant to which we agreed, upon the terms and subject to the conditions of the Meitav Purchase Agreement, to issue and sell to Meitav Dash in a private placement, certain of our securities in three tranches, as follows: (i) at the first closing, 9,500,000 ordinary shares, for a purchase price of NIS 3,800,000 ($1,013,874), (ii) at the second closing, 2,400,000 ordinary shares for a purchase price of NIS 960,000 ($256,136), provided that Meitav Dash shall not be obligated to buy or hold, immediately following the second closing, 20% or more of our share capital, and (iii) at the third closing for no additional consideration, warrants exercisable into 11,900,000 ordinary shares, or the Meitav Warrant.
Meitav Purchase Agreement
We completed the first and second closings on December 26, 2017 which resulted in the issuance to Meitav Dash of an aggregate of 11,900,000 ordinary shares for gross proceeds of NIS 4,760,000 ($1,270,010) and we completed the third closing on March 7, 2018 which resulted in the issuance to Meitav Dash of a warrant to purchase 11,900,000 ordinary shares represented by 238,000 ADSs.
The Meitav Purchase Agreement contains full-ratchet anti-dilution protection until the second anniversary of the first closing in the event of certain subsequent equity issuances at a price that is lower than the then applicable per ordinary share purchase price.
The Meitav Purchase Agreement also contains representations and warranties and covenants provisions customary in transactions of this nature.
Meitav Warrant
At the third closing, which we completed on March 7, 2018, we issued the Meitav Warrant exercisable into 11,900,000 ordinary shares, represented by 238,000 ADSs. The warrant may be exercised for a period of five years from issuance at an exercise price of the US dollar equivalent of NIS 40 per ADS (calculated in accordance with the known representative rate of exchange on the date of the notice of exercise).
The Meitav Warrant is subject to certain anti-dilution adjustments upon certain events, including share splits, share dividends, subsequent rights offerings, and fundamental transactions. In addition, pursuant to a side letter, the ordinary shares or ADSs issuable upon exercise of the Meitav Warrant are subject to full-ratchet anti-dilution protection until the second anniversary of the first closing in the event of certain subsequent equity issuances at a price that is lower than the then applicable per ordinary share purchase price.
Ami Sagy Financing
On November 9, 2017, we entered into the Sagy Purchase Agreement with Ami Sagy, pursuant to which we agreed, upon the terms and subject to the conditions of the Sagy Purchase Agreement, to issue and sell to Ami Sagy in a private placement, certain of our securities in two tranches, as follows: (i) at the first closing, 9,300,000 ordinary shares, for a purchase price of NIS 3,720,000 ($992,529), and (ii) at the second closing for no additional consideration, the Sagy Warrant exercisable into 9,300,000 ordinary shares.
Sagy Purchase Agreement
We completed the first closing on December 26, 2017 which resulted in the issuance to Ami Sagy of an aggregate of 9,300,000 ordinary shares for gross proceeds of NIS 3,720,000 ($992,529), and we completed the second closing on March 7, 2018 which resulted in the issuance to Ami Sagy of a warrant to purchase 9,300,000 ordinary shares represented by 186,000 ADSs.
130
The Sagy Purchase Agreement contains full-ratchet anti-dilution protection until the second anniversary of the first closing in the event of certain subsequent equity issuances at a price that is lower than the then applicable per ordinary share purchase price.
The Sagy Purchase Agreement also contains representations and warranties and covenants provisions customary in transactions of this nature.
Sagy Warrant
At the second closing, which we completed on March 7, 2018, we issued to Ami Sagy the Sagy Warrant exercisable into 9,300,000 ordinary shares. The Sagy Warrant may be exercised for a period of five years from issuance at an exercise price of the US dollar equivalent of NIS 40 per ADS (calculated in accordance with the known representative rate of exchange on the date of the notice of exercise).
The Sagy Warrant is subject to certain anti-dilution adjustments upon certain events, including share splits, share dividends, subsequent rights offerings, and fundamental transactions. In addition, pursuant to a side letter, the ordinary shares or ADSs issuable upon exercise of Sagy Warrant are subject to full-ratchet anti-dilution protection until the second anniversary of the first closing in the event of certain subsequent equity issuances at a price that is lower than the then applicable per ordinary share purchase price.
January 2018 Financing
On January 18, 2018, we entered into Security Purchase Agreements for the purchase and sale, in a private placement, of an aggregate of 4,344,340 ordinary shares for an aggregate of NIS 2,172,170 ($579,554) to the following three investors as follows: (i) Alpha entered into a Security Purchase Agreement for the purchase and sale of 1,275,340 ordinary shares for NIS 637,670 ($170,136); (ii) Ami Sagy entered into a Security Purchase Agreement for the purchase and sale of 2,046,000 ordinary shares for NIS 1,023,000 ($272,946); and (iii) Docor International BV entered into a Security Purchase Agreement for the purchase and sale of 1,023,000 ordinary shares for NIS 511,500 ($136,473). Closing occurred on January 25, 2018.
July 2018 Financing
On July 26, 2018, we entered into a Securities Purchase Agreement with Ami Sagy for the purchase and sale, in a private placement, of 11,125,000 ordinary shares for an aggregate purchase price of NIS 4,561,250 (approximately $1.2 million). Closing occurred on July 31, 2018.
131
| D. | Exchange Controls |
There are currently no Israeli currency control restrictions on remittances of dividends on our ordinary shares, proceeds from the sale of the shares or interest or other payments to non-residents of Israel, except for shareholders who are subjects of countries that are, or have been, in a state of war with Israel.
| E. | Taxation. |
The following description is not intended to constitute a complete analysis of all tax consequences relating to the acquisition, ownership and disposition of our ordinary shares and ADSs. You should consult your own tax advisor concerning the tax consequences of your particular situation, as well as any tax consequences that may arise under the laws of any state, local, foreign, or other taxing jurisdiction.
Israeli Tax Considerations and Government Programs
The following is a brief summary of the material Israeli tax laws applicable to us and certain Israeli Government programs that benefit us. This section also contains a discussion of material Israeli tax consequences concerning the ownership and disposition of our ordinary shares. This summary does not discuss all the aspects of Israeli tax law that may be relevant to a particular investor in light of his or her personal investment circumstances or to some types of investors subject to special treatment under Israeli law. Examples of such investors include residents of Israel or traders in securities who are subject to special tax regimes not covered in this discussion. To the extent that the discussion is based on new tax legislation that has not yet been subject to judicial or administrative interpretation, we cannot assure you that the appropriate tax authorities or the courts will accept the views expressed in this discussion. The discussion below is subject to change, including due to amendments under Israeli law or changes to the applicable judicial or administrative interpretations of Israeli law, which change could affect the tax consequences described below.
General Corporate Tax Structure in Israel
Israeli resident (as defined below) companies, such as us, are generally subject to corporate tax at the rate of 23% as of 2018. However, the effective tax rate payable by a company that derives income from a Preferred Enterprise or a Preferred Technology Enterprise (as discussed below) may be considerably lower. Capital gains derived by an Israeli company are generally subject to tax at the prevailing corporate tax rate.
Law for the Encouragement of Industry (Taxes), 5729-1969
The Law for the Encouragement of Industry (Taxes), 5729-1969, or the Industry Encouragement Law, provides several tax benefits for “Industrial Companies.”
The Industry Encouragement Law defines an “Industrial Company” as a company resident in Israel, of which 90% or more of its income in any tax year, other than income from defense loans, is derived from an “Industrial Enterprise” owned by it. An “Industrial Enterprise” is defined as an enterprise whose principal activity in a given tax year is industrial production.
The following corporate tax benefits, among others, are available to Industrial Companies:
| ● | amortization over an eight-year period of the cost of patents and rights to use a patent and know-how which were purchased in good faith and are used for the development or advancement of the Industrial Enterprise; |
| ● | deduction over a three-year period of expenses incurred in connection with the issuance and listing of shares on a stock market; and |
| ● | under certain conditions, an election to file consolidated tax returns with related Israeli Industrial Companies. |
There can be no assurance that we currently qualify, or will continue to qualify, as an Industrial Company or that the benefits described above will be available in the future.
132
Law for the Encouragement of Capital Investments, 5719-1959
Tax Benefits for Income from Preferred Enterprise
The Law for the Encouragement of Capital Investments, 5719-1959, or the Investment Law, currently provides certain tax benefits for income generated by “Preferred Companies” from their “Preferred Enterprises.” The definition of a Preferred Company includes, inter alia, a company incorporated in Israel that is not wholly owned by a governmental entity, which:
| ● | owns a Preferred Enterprise, which is defined as an “Industrial Enterprise” (as defined under the Investment Law) that is classified as either a “Competitive Enterprise” (as defined under the Investment Law) or a “Competitive Enterprise in the Field of Renewable Energy” (as defined under the Investment Law); |
| ● | is controlled and managed from Israel; |
| ● | is not a “Family Company,” a “Home Company,” or a “Kibbutz” (collective community) as defined under the Income Tax Ordinance; |
| ● | keeps acceptable books of account and files reports in accordance with the provisions of the Investment Law and the Income Tax Ordinance; and |
| ● | was not, and certain officers of which were not, convicted of certain crimes in the 10 years prior to the tax year with respect to which benefits are being claimed. |
As of January 1, 2017, a Preferred Company is currently entitled to a reduced corporate tax rate of 16% with respect to its income derived by its Preferred Enterprise, unless the Preferred Enterprise is located in development area A, in which case the rate is currently 7.5% (our operations are currently not located in development area A).
Dividends paid out of income attributed to a Preferred Enterprise are generally subject to tax at the rate of 20% or such lower rate as may be provided in an applicable tax treaty. However, if such dividends are paid to an Israeli company, such dividends should be exempt from tax (although, if such dividends are subsequently distributed to individuals or a non-Israeli company, tax at a rate of 20% or such lower rate as may be provided in an applicable tax treaty will apply).
If in the future we generate taxable income, to the extent that we qualify as a “Preferred Company,” the benefits provided under the Investment Law could potentially reduce our corporate tax liabilities. Therefore, the termination or substantial reduction of the benefits available under the Investment Law could materially increase our tax liabilities.
Tax Benefits for Income from Preferred Technology Enterprise
An amendment to the Investment Law was enacted as part of the Economic Efficiency Law that was published on December 29, 2016, and became effective as of January 1, 2017 (and is referred to herein as the “2017 Amendment”). The 2017 Amendment provides new tax benefits to Preferred Companies for “Technology Enterprises,” as described below, and is in addition to the Preferred Enterprise regime provided under the Investment Law.
133
The 2017 Amendment provides that a technology company satisfying certain conditions will qualify as a “Preferred Technology Enterprise” and may thereby enjoy a reduced corporate tax rate of 12% on income that qualifies as “Preferred Technology Income,” as defined in the Investment Law. The tax rate is further reduced to 7.5% for a Preferred Technology Enterprise located in development area A. In addition, a Preferred Technology Enterprise may enjoy a reduced capital gains tax rate of 12% on capital gain derived from the sale of certain “Benefited Intangible Assets” (as defined in the Investment Law) to a related foreign company if the Benefited Intangible Assets were acquired from a foreign company on or after January 1, 2017 for at least NIS 200 million, and the sale receives prior approval from the IIA.
Dividends distributed by a Preferred Technology Enterprise that are paid out of Preferred Technology Income are subject to tax at the rate of 20%, but if they are distributed to a foreign company and at least 90% of the shares of the distributing company are held by foreign resident companies then the tax rate may be as low as 4%, subject to the fulfillment of certain conditions.
As we have not yet generated taxable income, there is no assurance that we qualify as a Preferred Technology Enterprise or that the benefits described above will be available to us in the future.
If in the future we generate taxable income, to the extent that we qualify as a “Preferred Company,” the benefits provided under the Investment Law could potentially reduce our corporate tax liabilities. Therefore, the termination or substantial reduction of the benefits available under the Investment Law could materially increase our tax liabilities.
The Encouragement of Research, Development and Technological Innovation in the Industry Law 5744
Under the Encouragement of Research, Development and Technological Innovation in the Industry Law 5744-1984 (formerly known as the Law for the Encouragement of Research and Development in Industry 5744-1984), or Innovation Law, and the regulations and guidelines promulgated thereunder, research and development programs which meet specified criteria and are approved by a committee of the IIA, are eligible for grants. The grants awarded are typically up to 50% of the project’s expenditures, as determined by the research committee. The grantee is required to pay royalties to the State of Israel from the sale of products developed under the program. Regulations under the Innovation Law generally provide for the payment of royalties of 3% to 6% on income generated from products and services based on technology developed using grants, until 100% of the grant, linked to the dollar and bearing interest at the LIBOR rate, is repaid. In July 2017, new regulations came into force. According to the new regulations, the royalties range between 1.3-5% depending on the company’s size and sector. The terms of the IIA participation also require that products developed with IIA grants be manufactured in Israel and that the know-how developed thereunder may not be transferred outside of Israel, unless approval is received from the IIA and additional payments are made to the IIA. However, this does not restrict the export of products that incorporate the funded know-how. The royalty repayment ceiling can reach up to three times the amount of the grant received (plus interest) if manufacturing is transferred outside of Israel, and repayment of up to six times the amount of the grant (plus interest) may be required if the technology itself is transferred outside of Israel or license to use it was granted to a foreign entity.
Taxation of our Shareholders
Capital Gains Tax
Israeli law generally imposes a capital gains tax (i) on the sale of any capital assets by residents of Israel, as defined for Israeli tax purposes, and (ii) on the sale of capital assets located in Israel, including shares of Israeli companies, by non-residents of Israel, unless a specific exemption is available or unless a tax treaty between Israel and the shareholder’s country of residence provides otherwise. The law distinguishes between real gain and inflationary surplus. The inflationary surplus is a portion of the total capital gain that is equivalent to the increase of the relevant asset’s purchase price which is attributable to the increase in the Israeli consumer price index or a foreign currency exchange rate between the date of purchase and the date of sale. The real gain is the excess of the total capital gain over the inflationary surplus.
134
Israeli Residents
Generally, as of January 1, 2012 and thereafter, the tax rate applicable to real capital gains derived from the sale of shares, whether listed on a stock market or not, is 25% for Israeli individuals, unless such shareholder claims a deduction for financing expenses in connection with such shares, in which case the gain will generally be taxed at a rate of 30%. Additionally, if such shareholder is considered a “substantial shareholder” at the time of the sale or at any time during the 12-month period preceding such sale, the tax rate will be 30%. A “substantial shareholder” is defined as one who holds, directly or indirectly, alone or “together with another” (i.e., together with a relative, or together with someone who is not a relative but with whom, according to an agreement, there is regular cooperation in material matters of the company, directly or indirectly), holds, directly or indirectly, at least 10% of any of the “means of control” in the company. “Means of control” generally include the right to vote, receive profits, nominate a director or an executive officer, receive assets upon liquidation, or instruct someone who holds any of the aforementioned rights regarding the manner in which such rights are to be exercised. However, different tax rates will apply to dealers in securities. Israeli companies are subject to capital gains tax at the regular corporate tax rate (i.e., 23% for the tax year 2018 and thereafter) on real capital gains derived from the sale of listed shares.
As of January 1, 2019, Israeli resident shareholders who are individuals with taxable income that exceeds NIS 649,500 in a tax year (linked to the Israeli consumer price index each year) will be subject to an additional tax at the rate of 3% on the portion of their taxable income for such tax year that is in excess of NIS 649,500 (linked to the Israeli consumer price index each year). For this purpose, taxable income includes taxable capital gains from the sale of our shares and taxable income from dividend distributions.
In some instances where our shareholders are liable for Israeli tax on the sale of their ordinary shares, the payment of the consideration may be subject to the withholding of Israeli tax at source.
Non-Israeli Residents
A non-Israeli resident who derives capital gains from the sale of shares in an Israeli resident company that were purchased after the company was listed for trading on a stock exchange outside of Israel will be exempt from Israeli tax so long as the shares were not held through a permanent establishment that the non-resident maintains in Israel. However, non-Israeli resident corporations will not be entitled to the foregoing exemption if (i) an Israeli resident has a controlling interest, directly or indirectly, alone, “together with another” (as defined above), or together with another Israeli resident, of more than 25% in one or more of the “means of control” (as defined above) in such non-Israeli resident corporation, or (ii) Israeli residents are the beneficiaries of, or are entitled to, 25% or more of the revenues or profits of such non-Israeli resident corporation, whether directly or indirectly.
In addition, a sale of securities by a non-Israeli resident may be exempt from Israeli capital gains tax under the provisions of an applicable tax treaty. For example, pursuant to the provisions of the Convention between the Government of the United States of America and the Government of the State of Israel with respect to Taxes on Income, as amended, or the U.S.-Israel Tax Treaty, capital gains arising from the sale, exchange or disposition of our ordinary shares by (i) a person who qualifies as a resident of the United States within the meaning of the U.S.-Israel Tax Treaty, (ii) who holds the shares as a capital asset, and (iii) who is entitled to claim the benefits afforded to such person by the U.S.-Israel Tax Treaty generally is generally exempt from Israeli capital gains tax. Such exemption will not apply if: (i) such person holds, directly or indirectly, shares representing 10% or more of our voting power during any part of the 12-month period preceding such sale, exchange, or disposition, subject to particular conditions; (ii) the capital gains from such sale, exchange, or disposition are attributable to a permanent establishment in Israel; or (iii) such person is an individual and was present in Israel for 183 days or more during the relevant tax year. In such case, the capital gain arising from the sale, exchange, or disposition of our ordinary shares would be subject to Israeli tax, to the extent applicable; however, under the U.S.-Israel Tax Treaty, the taxpayer may be permitted to claim a credit for such taxes against the U.S. federal income tax imposed with respect to such sale, exchange, or disposition, subject to the limitations under U.S. law applicable to foreign tax credits. The U.S.-Israel Tax Treaty does not relate to U.S. state or local taxes.
Shareholders may be required to demonstrate that they are exempt from tax on their capital gains in order to avoid withholding at source at the time of sale.
It should be noted that in the event that the real capital gain realized by an individual shareholder is not exempt from tax in Israel, the tax rates applicable to Israeli resident individual shareholders should generally apply.
In some instances where our shareholders may be liable for Israeli tax on the sale of their ordinary shares, the payment of the consideration may be subject to the withholding of Israeli tax at source.
135
Taxation of Dividend Distributions
Israeli Residents
Israeli resident individuals are generally subject to Israeli income tax on the receipt of dividends paid on our ordinary shares, other than bonus shares (share dividends). As of January 1, 2012 and thereafter, the tax rate applicable to such dividends is generally 25%. With respect to a person who is a “substantial shareholder” (as defined above) at the time the dividend is received or at any time during the preceding 12-month period, the applicable tax rate is 30%. Dividends paid from income derived from Preferred Enterprises and Preferred Technology Enterprises will generally be subject to income tax at a rate of 20%.
As of January 1, 2019, Israeli resident shareholders who are individuals with taxable income that exceeds NIS 649,500 in a tax year (linked to the Israeli consumer price index each year) will be subject to an additional tax at the rate of 3% on the portion of their taxable income for such tax year that is in excess of NIS 649,500 (linked to the Israeli consumer price index each year). For this purpose, taxable income includes taxable capital gains from the sale of our shares and taxable income from dividend distributions.
Dividends paid to an Israeli resident individual shareholder on our ordinary shares will generally be subject to withholding tax at the rates corresponding with the income tax rates detailed above unless we are provided in advance with a withholding tax certificate issued by the Israel Tax Authority stipulating a different rate.
Notwithstanding the above, dividends paid to an Israeli resident “substantial shareholder” (as defined above) on publicly traded shares, like our ordinary shares, which are held via a “nominee company” (as defined under the Israeli Securities Law), are generally subject to Israeli withholding tax at a rate of 25%, unless a different rate is provided under an applicable tax treaty, provided that a certificate from the Israel Tax Authority allowing for a reduced withholding tax rate is obtained in advance.
If the dividend is attributable partly to income derived from a Preferred Enterprise or a Preferred Technology Enterprise and partly to other sources of income, the tax rate will be a blended rate reflecting the relative portions of the various types of income. We cannot assure you that we will designate the profits that are being distributed in a way that will reduce shareholders’ tax liability.
Israeli resident companies are generally exempt from tax on the receipt of dividends paid on our ordinary shares.
Non-Israeli Residents
Unless relief is provided in a treaty between Israel and the shareholder’s country of residence, non-Israeli residents are generally subject to Israeli income tax on the receipt of dividends paid on our ordinary shares at the rate of 25%. With respect to a person (including a corporation) who is a “substantial shareholder” (as defined above) at the time of receiving the dividend or at any time during the preceding 12-month period, absent treaty relief as mentioned above, the applicable Israeli income tax rate is 30%. Notwithstanding the above, dividends paid from income derived from Preferred Enterprises will be subject to Israeli income tax at a rate of 20%. In addition, dividends distributed by a Preferred Technology Enterprise that are paid out of Preferred Technology Income are subject to tax at the rate of 20%, but if they are distributed to a foreign company and at least 90% of the shares of the distributing company are held by foreign resident companies then the tax rate may be as low as 4%, subject to the fulfillment of certain conditions.
In this regard, dividends paid to a non-Israeli resident shareholder on our ordinary shares will generally be subject to withholding tax at the rates corresponding with the income tax rates detailed above unless we are provided in advance with a withholding tax certificate issued by the Israel Tax Authority stipulating a different rate (e.g., in accordance with the provisions of an applicable tax treaty).
Notwithstanding the above, dividends paid to a non-Israeli resident “substantial shareholder” (as defined above) on publicly traded shares, like our ordinary shares, which are held via a “nominee company” (as defined under the Israeli Securities Law), are generally subject to Israeli withholding tax at a rate of 25%, unless a different rate is provided under an applicable tax treaty, provided that a certificate from the Israel Tax Authority allowing for a reduced withholding tax rate is obtained in advance.
136
In addition, it should be noted that an additional 3% tax might be applicable to individual shareholders if certain conditions are met.
Under the U.S.-Israel Tax Treaty, the maximum Israeli tax on dividends paid to a holder of ordinary shares who qualifies as a resident of the United States within the meaning of the U.S.-Israel Tax Treaty is 25%. Such tax rate is generally reduced to 12.5% if: (i) the shareholder is a U.S. corporation and holds at least 10% of the outstanding shares of our voting stock during the part of our tax year that precedes the date of payment of the dividends and during the whole of our prior tax year; (ii) not more than 25% of our gross income in the tax year preceding the payment of the dividends consists of interest or dividends, other than dividends or interest received from subsidiary corporations 50% or more of the outstanding shares of voting stock of which is owned by us at the time such dividends or interest are received by us; and (iii) the dividends are not sourced from income derived during a period for which we were entitled to the reduced tax rate applicable to a Preferred Enterprise under the Investment Law. If the dividends are sourced from income derived during a period for which we are entitled to the reduced tax rate applicable to a Preferred Enterprise or a Preferred Technology Enterprise under the Investment Law, to the extent that the first two conditions detailed above are met, the Israeli tax rate applicable to such dividends should be 15%.
If the dividend is attributable partly to income derived from a Preferred Enterprise or a Preferred Technology Enterprise and partly to other sources of income, the tax rate will be a blended rate reflecting the relative portions of the various types of income. We cannot assure you that we will designate the profits that are being distributed in a way that will reduce shareholders’ tax liability.
Estate and gift tax
Israeli law presently does not impose estate tax.
Israeli law also does not presently impose gift taxes upon the transfer of assets to Israeli resident individuals so long as it is demonstrated to the satisfaction of the Israel Tax Authority that the transfer was executed in good faith.
Material U.S. Federal Income Tax Consequences
The following summary describes certain material U.S. federal income tax consequences relating to an investment in the ADSs and ordinary shares. This summary deals only with ADSs and ordinary shares that are held as capital assets within the meaning of section 1221 of the U.S. Internal Revenue Code of 1986, as amended, or the Code, and does not address tax considerations of holders that may be subject to special tax rules, such as dealers or traders in securities or currencies, financial institutions, tax-exempt organizations, insurance companies, regulated investment companies, real estate investment trusts, individual retirement and tax-deferred accounts, persons holding ADSs or ordinary shares as part of a hedging, integrated, conversion or constructive sale transaction, or a straddle, persons subject to the alternative minimum tax, or persons who have a functional currency other than the U.S. dollar. In addition, this discussion does not address the tax treatment of U.S. holders (as defined below) who own, directly, indirectly, or constructively, 10% or more of our outstanding stock, by vote or value. The summary set forth below relating to U.S. holders (as defined below) is applicable only to such U.S. holders (i) who are residents of the United States for purposes of the United States-Israel Tax Treaty, (ii) whose ordinary shares or ADSs are not, for purposes of the United States-Israel Tax Treaty, effectively connected with or attributable to a permanent establishment in Israel, and (iii) who otherwise qualify for the full benefits of the United States-Israel Tax Treaty. The discussion below is based upon the Code, final, temporary and proposed Treasury regulations promulgated thereunder, applicable administrative rulings and judicial interpretations thereof, and the United States-Israel Tax Treaty, all as in effect as of the date hereof and all of which are subject to change, possibly on a retroactive basis, and all of which are open to differing interpretations. In addition, this summary does not consider the possible application of U.S. federal gift or estate taxes or any aspect of state, local, or non-U.S. tax laws. Furthermore, we will not seek a ruling from the IRS with regard to the U.S. federal income tax treatment of an investment in our ADSs or ordinary shares and can provide no assurance that the tax consequences contained in this summary will not be challenged by the IRS or will be sustained in a court if challenged.
137
As used in this summary the term “U.S. holder” means a beneficial owner of ADSs or ordinary shares that is, for U.S. federal income tax purposes: (i) an individual citizen or resident of the United States, (ii) a corporation (or other entity taxable as a corporation for U.S. federal income tax purposes) created or organized in or under the laws of the United States or any political subdivision thereof, (iii) an estate the income of which is subject to U.S. federal income taxation regardless of its source, or (iv) a trust if either (a) a court within the United States is able to exercise primary supervision over the administration of the trust and one or more U.S. persons have the authority to control all substantial decisions of the trust, or (b) the trust has a valid election in effect under applicable Treasury regulations to be treated as a U.S. person. Except to the limited extent discussed below, this summary does not consider the U.S. federal tax considerations to a person that is not a U.S. holder (a “non-U.S. holder”). In addition, the tax treatment of persons who hold ADSs or ordinary shares through a partnership or other pass-through entity treated as a partnership for U.S. federal income tax purposes generally depends upon the status of the partner and the activities of the partnership. The tax consequences to such a partner or partnership are not considered in this summary and partners and partnerships should consult their tax advisors with respect to the U.S. federal tax consequences of investing in the ADSs or ordinary shares.
This summary does not discuss all aspects of U.S. federal income taxation that may be relevant to a particular investor in light of its circumstances. Prospective purchasers of the ADSs or ordinary shares should consult their own tax advisors with respect to the specific U.S. federal income tax consequences to such person of purchasing, holding, or disposing of the ADSs or ordinary shares, as well as the effect of any state, local, or other tax laws.
ADSs
If you hold ADSs, for U.S. federal income tax purposes, you generally will be treated as the owner of the underlying ordinary shares that are represented by such ADSs. Accordingly, deposits or withdrawals of ordinary shares for ADSs will not be subject to U.S. federal income tax.
Distributions on Ordinary Shares or ADSs
Subject to the discussion under the heading “Passive Foreign Investment Company Consequences,” U.S. holders are required to include in gross income the amount of any distribution paid on ordinary shares or ADSs to the extent the distribution is paid out of our current and/or accumulated earnings and profits, as determined for U.S. federal income tax purposes. To the extent a distribution paid with respect to our ordinary shares or ADSs exceeds our current and accumulated earnings and profits, such amount will be treated first as a non-taxable return of capital, reducing a U.S. holder’s tax basis for the ordinary shares or ADSs to the extent thereof, and thereafter as either long-term or short-term capital gain depending upon whether the U.S. holder has held our ordinary shares or ADSs for more than one year as of the time such distribution is received. Preferential tax rates for long-term capital gains are applicable for U.S. holders that are individuals, estates, or trusts. However, we do not expect to maintain calculations of our earnings and profits under United States federal income tax principles. Therefore, U.S. holders should expect that the entire amount of any distribution generally will be reported as dividend income. The amount of the dividend will generally be treated as foreign-source dividend income to U.S. holders. A non-corporate U.S. holder that meets certain eligibility requirements may qualify for a lower rate of U.S. federal income taxation on dividends paid if we are a “qualified foreign corporation” for U.S. federal income tax purposes. We generally will be treated as a qualified foreign corporation if we are not a passive foreign investment company, or PFIC, in the taxable year in which such dividends are paid or in the preceding taxable year (see discussion below), and (i) we are eligible for benefits under the United States-Israel income tax treaty or (ii) our ordinary shares or ADSs are listed on an established securities market in the United States (which includes the Nasdaq Capital Market). In addition, a non-corporate U.S. holder will not be eligible for a reduced U.S. federal income tax rate with respect to dividend distributions on ordinary shares or ADSs if (a) such U.S. holder has not held the ordinary shares or ADSs for at least 61 days during the 121-day period starting on the date which is 60 days before, and ending 60 days after the ex-dividend date, (b) to the extent the U.S. holder is under an obligation to make related payments on substantially similar or related property, or (c) with respect to any portion of a dividend that is taken into account by the U.S. holder as investment income under Section 163(d)(4)(B) of the Code. Any days during which the U.S. holder has diminished its risk of loss with respect to ordinary shares or ADSs (for example, by holding an option to sell the ordinary shares or ADSs) are not counted towards meeting the 61-day holding period. Non-corporate U.S. holders should consult their own tax advisors concerning whether dividends received by them qualify for the reduced rate of tax.
Corporate U.S. holders generally will not be allowed a deduction for dividends received from us.
138
The amount of a distribution with respect to our ordinary shares or ADSs equals the amount of cash and the fair market value of any property distributed plus the amount of any Israeli taxes withheld therefrom. The amount of any cash distributions paid in NIS equals the U.S. dollar value of the NIS on the date of distribution based upon the exchange rate in effect on such date, regardless of whether the NIS are converted into U.S. dollars at that time, and U.S. holders who include such distribution in income on such date will have a tax basis in such NIS for U.S. federal income tax purposes equal to such U.S. dollar value. If the dividend is converted to U.S. dollars on the date of receipt, a U.S. holder generally will not recognize a foreign currency gain or loss. However, if the U.S. holder converts the NIS into U.S. dollars on a later date, the U.S. holder must include, in computing its income, any gain or loss resulting from any exchange rate fluctuations. The gain or loss will be equal to the difference between (i) the U.S. dollar value of the amount included in income when the dividend was received and (ii) the amount received on the conversion of the NIS into U.S. dollars. Such gain or loss will generally be ordinary income or loss and United States source income for U.S. foreign tax credit purposes. U.S. holders should consult their own tax advisors regarding the tax consequences to them if we pay dividends in NIS or any other non-U.S. currency.
Subject to certain significant conditions and limitations, including potential limitations under the U.S.-Israel Tax Treaty, U.S. holders may be entitled to a credit against their U.S. federal income tax liability or a deduction against U.S. federal taxable income in an amount equal to the Israeli tax withheld on distributions on our ordinary shares or ADSs. U.S. holders should consult their own tax advisors to determine whether and to what extent they would be entitled to such credit. Distributions paid on our ordinary shares or ADSs will generally be treated as passive income that is foreign source for U.S. foreign tax credit purposes, which may be relevant in calculating a U.S. holder’s foreign tax credit limitation.
Disposition of Ordinary Shares or ADSs
Subject to the discussion under the heading “Passive Foreign Investment Company Consequences,” upon the sale, exchange or other disposition ordinary shares or of ADSs, a U.S. holder generally will recognize capital gain or loss in an amount equal to the difference between the amount realized on the disposition and such U.S. holder’s adjusted tax basis in the ordinary shares or ADSs. The adjusted tax basis in an ordinary share or ADS generally will be equal to the cost of such ordinary share or ADS. The capital gain or loss realized on the sale, exchange, or other disposition of ordinary shares or ADSs will be long-term capital gain or loss if the U.S. holder held the ordinary shares or ADSs for more than one year as of the time of disposition. Preferential tax rates for long-term capital gain will generally apply to non-corporate U.S. holders. Any gain or loss realized by a U.S. holder on the sale, exchange, or other disposition of ordinary shares or ADSs generally will be treated as from sources within the United States for U.S. foreign tax credit purposes, except for certain losses which will be treated as foreign source to the extent certain dividends were received (or certain inclusion amounts were taken into account) by the U.S. holder within the 24-month period preceding the date on which the U.S. holder recognized the loss. The deductibility of capital losses for U.S. federal income tax purposes is subject to limitations.
Disclosure of Reportable Transactions
If a U.S. holder sells or disposes of the ordinary shares or ADSs at a loss or otherwise incurs certain losses that meet certain thresholds, such U.S. holder may be required to file a disclosure statement with the IRS. Failure to comply with these and other reporting requirements could result in the imposition of significant penalties.
Passive Foreign Investment Company Consequences
Generally, a non-U.S. corporation will be a PFIC for U.S. federal income tax purposes in any taxable year in which either (i) 75% or more of its gross income for such year consists of certain types of “passive” income or (ii) 50% or more of the average fair market value of its assets during such year (based on quarterly valuations) produce or are held for the production of passive income. Passive income for this purpose generally includes dividends, interest, rents, royalties, annuities, income from certain commodities transactions and from notional principal contracts, and the excess of gains over losses from the disposition of assets that produce passive income. Passive income also includes amounts derived by reason of the temporary investment of funds, including those raised in a public offering. Assets that produce or are held for the production of passive income include cash, even if held as working capital or raised in a public offering, marketable securities, and other assets that may produce passive income. In determining whether a non-U.S. corporation is a PFIC, a proportionate share of the income and assets of each corporation in which it owns, directly or indirectly, at least a 25% interest (by value) is taken into account.
139
A foreign corporation’s PFIC status is an annual determination that is based on tests that are factual in nature, and our PFIC status for any year will depend on the composition of our income, fair market value of our assets, and our activities for such year. Based on our non-passive revenue-producing operations for the year ended December 31, 2018, we do not expect to be a PFIC for our 2018 taxable year. Because the PFIC determination is highly fact intensive, there can be no assurance that we will not be a PFIC in 2019 or any other year. Even if we determine that we are not a PFIC after the close of a taxable year, there can be no assurance that the IRS or a court will agree with our conclusion.
If we were a PFIC for any taxable year during which a U.S. holder held ordinary shares or ADSs, then unless an election has been made by a U.S. holder to be taxed under one of the alternative regimes discussed below, gain recognized by a U.S. holder on a sale or other disposition (including certain pledges) of the ordinary shares or ADSs would be allocated ratably over the U.S. holder’s holding period for the ordinary shares or ADSs. The amounts allocated to the taxable year of the sale or other disposition and to any year before we became a PFIC would be taxed as ordinary income. The amount allocated to each other taxable year would be subject to tax at the highest rate in effect for individuals or corporations, as appropriate, for that taxable year, and an interest charge would be imposed on the amount allocated to that taxable year. Similar rules would apply to any distribution with respect to the ordinary shares or ADSs in excess of 125% of the average of the annual distributions received by a U.S. holder during the preceding three years or such U.S. holder’s holding period, whichever is shorter. In addition, non-corporate U.S. holders will not be eligible for reduced rates of taxation on any dividends received from us if we are a PFIC in the taxable year in which such dividends are paid or in the preceding taxable year.
If we are a PFIC for any taxable year during which you hold the ordinary shares or ADSs and our non-United States subsidiary is also a PFIC, a U.S. holder would be treated as owning a proportionate amount (by value) of the shares of the lower-tier PFIC for purposes of the application of these rules. U.S. holders are urged to consult their tax advisors about the application of the PFIC rules to our subsidiary.
If we are treated as a PFIC for any taxable year during the holding period of a non-electing U.S. holder (i.e., a U.S. holder that does not elect to be taxed under one of the alternative regimes discussed below), we will continue to be treated as a PFIC for all succeeding years during which such non-electing U.S. holder is treated as a direct or indirect holder even if we are not a PFIC for such years. A U.S. holder is encouraged to consult its tax advisor with respect to any available elections that may be applicable in such a situation, including the “deemed sale” election of Section 1298(b)(1) of the Code.
Notwithstanding the default PFIC rules described in the preceding paragraphs, certain elections may be available that would result in alternative tax consequences; i.e., the “qualified electing fund” or “QEF” election and the “mark to market” election. If a U.S. holder makes a timely and valid mark-to-market election, the U.S. holder generally will recognize as ordinary income any excess of the fair market value of the ordinary shares or ADSs at the end of each taxable year over their adjusted tax basis, and will recognize an ordinary loss in respect of any excess of the adjusted tax basis of the ordinary shares or ADSs over their fair market value at the end of the taxable year (but only to the extent of the net amount of income previously included as a result of the mark-to-market election). The U.S. holder’s tax basis in the ordinary shares or ADSs will be adjusted to reflect the income or loss resulting from the mark-to-market election. Any gain recognized on the sale or other disposition of ordinary shares or ADSs in a year when we are a PFIC will be treated as ordinary income and any loss will be treated as an ordinary loss (but only to the extent of the net amount of income previously included as a result of the mark-to-market election and any loss in excess of such amount will be treated as capital loss). The mark-to-market election is available only if we are a PFIC and the ordinary shares or ADSs are “regularly traded” on a “qualified exchange” within the meaning of applicable U.S. Treasury regulations. The ordinary shares or ADSs will be treated as “regularly traded” in any calendar year in which more than a de minimis quantity of the ordinary shares or ADSs are traded on a qualified exchange on at least 15 days during each calendar quarter. Although the IRS has not published any authority identifying specific exchanges that may constitute “qualified exchanges,” Treasury Regulations provide that a qualified exchange is (i) a U.S. securities exchange that is registered with the Securities and Exchange Commission, (ii) the U.S. market system established pursuant to section 11A of the Securities and Exchange Act of 1934, or (iii) a non-U.S. securities exchange that is regulated or supervised by a governmental authority of the country in which the market is located, provided that: (a) such non-U.S. exchange has trading volume, listing, financial disclosure, surveillance, and other requirements designed to prevent fraudulent and manipulative acts and practices, to remove impediments to and perfect the mechanism of a free and open, fair and orderly, market, and to protect investors, and the laws of the country in which such non-U.S. exchange is located and the rules of such non-U.S. exchange ensure that such requirements are actually enforced; and (b) the rules of such non-U.S. exchange effectively promote active trading of listed shares. No assurance can be given that the ADSs will meet the requirements to be treated as “regularly traded” for purposes of the mark-to-market election. The Nasdaq Capital Market is a qualified exchange for this purpose and, consequently, if the ADSs are regularly traded, the mark-to-market election will be available to a U.S. holder. We have delisted our ordinary shares from the Tel Aviv Stock Exchange and the last date of trading of our ordinary shares was on October 29, 2018. Therefore, U.S. holders are not currently able to make a mark-to-market election with respect to our ordinary shares. A mark-to-market election will not apply to ordinary shares or ADSs held by a U.S. holder for any taxable year during which we are not a PFIC, but will remain in effect with respect to any subsequent taxable year in which we become a PFIC unless the ordinary shares or ADSs are no longer regularly traded on a qualified exchange or the IRS consents to the revocation of the election. Such election will not apply to any PFIC subsidiary that we own. Each U.S. holder is encouraged to consult its own tax advisor with respect to the availability and tax consequences of a mark-to-market election with respect to the ordinary shares or ADSs.
140
Another way in which certain of the adverse consequences of PFIC status can be mitigated is for a U.S. holder to make a QEF election. Generally, a shareholder making the QEF election is required for each taxable year to include in income a pro rata share of the ordinary earnings and net capital gain of the QEF, subject to a separate election to defer payment of taxes, which deferral is subject to an interest charge. An election to treat us as a QEF will not be available if we do not provide the information necessary to make such an election. We are not obligated and do not currently intend to provide the information necessary to make a QEF election and thus it is not expected that a QEF election will be available for U.S. holders of the ordinary shares or ADSs if we were a PFIC in any prior year, the current year or any future year.
U.S. holders should consult their tax advisors to determine under what circumstances these elections would be available and, if available, what the consequences of the alternative treatments would be in their particular circumstances.
If a U.S. holder holds ordinary shares or ADSs in any year in which we are treated as a PFIC, the U.S. holder will be required to file IRS Form 8621 and may be subject to certain other information reporting requirements.
The U.S. federal income tax rules relating to PFICs are complex. Prospective U.S. holders are urged to consult their own tax advisors with respect to the consequences to them of an investment in a PFIC, any elections available with respect to the ADSs or ordinary shares and the IRS information reporting obligations with respect to the purchase, ownership, and disposition of the ADSs or ordinary shares in the event we are determined to be a PFIC.
Medicare Tax on Investment Income
In addition to the income taxes described above, U.S. holders that are individuals, estates, or trusts and whose income exceeds certain thresholds will be subject to a 3.8% tax on all or a portion of their “net investment income,” which generally results from dividends and dispositions of ordinary shares or ADSs. U.S. holders should consult their tax advisors with respect to the applicability of the 3.8% Medicare tax to their income and gains, if any, resulting from their investment in the ordinary shares or ADSs.
Information Reporting and Backup Withholding
A U.S. holder may be subject to backup withholding and information reporting requirements with respect to cash distributions and proceeds from a disposition of ADSs or ordinary shares. In general, backup withholding will apply only if a U.S. holder fails to comply with certain identification procedures. Information reporting and backup withholding will not apply with respect to payments made to certain exempt recipients, such as corporations and tax-exempt organizations. Backup withholding is not an additional tax and may be claimed as a credit against the U.S. federal income tax liability of a U.S. holder, provided that the required information is furnished to the IRS.
141
Tax Reporting
Certain U.S. holders will be required to file an IRS Form 926 (Return by a U.S. Transferor of Property to a Foreign Corporation) to report a transfer of cash or other property to us. Substantial penalties may be imposed on a U.S. holder that fails to comply with this reporting requirement. Each U.S. holder is urged to consult with its own tax advisor regarding this reporting obligation.
Foreign Asset Reporting
Certain U.S. holders who are individuals may be required to report information relating to an interest in the ADSs or ordinary shares, subject to certain exceptions. For example, individuals that own “specified foreign financial assets” with an aggregate value in excess of $50,000 are generally required to file IRS Form 8938 with respect to such assets with their tax returns. “Specified foreign financial assets” include any financial accounts maintained by foreign financial institutions, as well as any of the following, but only if they are not held in accounts maintained by financial institutions: (i) stocks and securities issued by non-U.S. persons; (ii) financial instruments and contracts held for investment that have non-U.S. issuers or counterparties; and (iii) interests in foreign entities. Certain domestic entities that are U.S. holders may also be required to file Form 8938 in the near future. In addition, a U.S. holder should consider the possible obligation to file FinCEN Form 114, Report of Foreign Bank and Financial Accounts, as a result of holding ADSs or ordinary shares. U.S. holders are urged to consult their tax advisors regarding the application of these and other reporting requirements that may apply to their ownership of ADSs or ordinary shares.
Non-U.S. Holders of Ordinary Shares or ADSs
Except as provided below, a non-U.S. holder of ordinary shares or ADSs generally will not be subject to U.S. income or withholding tax on the payment of dividends on and the proceeds from the disposition of ADSs or ordinary shares.
A non-U.S. holder may be subject to U.S. federal income tax on dividends received on ADSs or ordinary shares or upon the receipt of income from the disposition of ADSs or ordinary shares if: (i) such income is effectively connected with the conduct by the non-U.S. holder of a trade or business in the United States or, in the case of a resident of a country which has an applicable income tax treaty with the United States, such item is attributable to a permanent establishment or a fixed place of business of the non-U.S. holder in the United States; (ii) with respect to a U.S. holder that is an individual, the non-U.S. holder is an individual who is present in the United States for 183 days or more in the taxable year of the sale and certain other conditions are met; or (iii) the non-U.S. holder is subject to tax pursuant to the provisions of the U.S. tax laws applicable to U.S. expatriates.
Payments to non-U.S. holders of distributions on, or proceeds from the disposition of, ADSs or ordinary shares are generally exempt from information reporting and backup withholding. However, a non-U.S. holder may be required, under certain circumstances, to establish that exemption by providing certification of non-U.S. status on an appropriate IRS Form W-8.
THE DISCUSSION ABOVE IS A GENERAL SUMMARY AND IS NOT INTENDED TO CONSTITUTE A COMPLETE ANALYSIS OF ALL TAX CONSEQUENCES RELATING TO THE PURCHASE, OWNERSHIP AND DISPOSITION OF THE ADSs OR ORDINARY SHARES. IT DOES NOT COVER ALL TAX MATTERS THAT MAY BE OF IMPORTANCE TO A PROSPECTIVE INVESTOR. EACH PROSPECTIVE INVESTOR IS URGED TO CONSULT ITS OWN TAX ADVISOR ABOUT THE TAX CONSEQUENCES TO IT RELATING TO THE PURCHASE, OWNERSHIP, AND DISPOSITION OF ADSs OR ORDINARY SHARES IN LIGHT OF THE INVESTOR’S OWN CIRCUMSTANCES.
| F. | Dividends and Paying Agents |
Not applicable.
142
| G. | Statement by Experts |
Not applicable.
| H. | Documents on Display |
We are subject to certain information reporting requirements of the Exchange Act, applicable to foreign private issuers and under those requirements will file reports with the SEC. The SEC maintains an internet site at http://www.sec.gov that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC. Our filings with the SEC will also available to the public through the SEC’s website at www.sec.gov.
As a foreign private issuer, we are exempt from the rules under the Exchange Act related to the furnishing and content of proxy statements, and our officers, directors and principal shareholders will be exempt from the reporting and short-swing profit recovery provisions contained in Section 16 of the Exchange Act. In addition, we are not required under the Exchange Act to file annual, quarterly and current reports and financial statements with the SEC as frequently or as promptly as U.S. domestic companies whose securities are registered under the Exchange Act. However, we will file with the SEC, within 120 days after the end of each fiscal year, or such applicable time as required by the SEC, an annual report on Form 20-F containing financial statements audited by an independent registered public accounting firm, and may submit to the SEC, on a Form 6-K, unaudited quarterly financial information.
| I. | Subsidiary Information. |
Not applicable.
| ITEM 11. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
We are exposed to market risk in the ordinary course of our business. Market risk represents the risk of loss that may impact our financial position due to adverse changes in financial market prices and rates. Our market risk exposure is primarily a result of fluctuations in foreign currency exchange rates and interest rates.
Foreign Currency Exchange Risk
Our functional and reporting currency is the New Israeli Shekel (NIS) which is the local currency in Israel. Our foreign currency exposures give rise to market risk associated with exchange rate movements of the NIS, mainly against the U.S. dollar and the Euro. Although the NIS is our functional currency, a small portion of our expenses consist principally of payments made to subcontractors and consultants for clinical trials, other research and development activities, and purchase of new equipment. A material portion of our research and development is conducted through collaboration agreements denominated in U.S. dollars, and therefore our net research and development expenses are subject to significant foreign currency risk. If the NIS fluctuates significantly against either the U.S. dollar or the Euro, it may have a negative impact on our results of operations. To date, such fluctuations in exchange rates have not materially affected our results of operations or financial condition for the periods under review.
To date, we have not entered into any hedging arrangements with respect to foreign currency risk or other derivative financial instruments. In the future, we may enter into currency hedging transactions to decrease the risk of financial exposure from fluctuations in the operating currencies. These measures, however, may not adequately protect us from the material adverse effects of such fluctuations.
143
Interest Rate Risk
At present, our investments consist primarily of cash and cash equivalents in short-term deposits. The primary objective of our investment activities is to preserve our capital to fund our operations. Our investments are exposed to market risk due to fluctuation in interest rates, which may affect our interest income and the fair market value of our investments, if any. We manage this exposure by performing ongoing evaluations of our investments. Due to the short-term maturities, if any, of our investments to date, their carrying value has always approximated their fair value. We believe that our exposure to interest rate risk is not significant and a 1% change in market interest rates would not have a material impact on our assets.
| ITEM 12. | DESCRIPTION OF SECURITIES OTHER THAN EQUITY SECURITIES |
| A. | Debt Securities. |
Not applicable.
| B. | Warrants and rights. |
Not applicable.
| C. | Other Securities. |
Not applicable.
| D. | American Depositary Shares |
| Persons depositing or withdrawing ordinary shares or ADS holders must pay: | For: | |
| $5.00 (or less) per ADSs (or portion of ADSs) | Issuance of ADSs, including issuances resulting from a distribution of ordinary shares or rights or other property; or cancellation of ADSs for the purpose of withdrawal, including if the deposit agreement terminates | |
| $0.05 (or less) per ADS | Any cash distribution to ADS holders | |
| A fee equivalent to the fee that would be payable if securities distributed to you had been ordinary shares and the ordinary shares had been deposited for issuance of ADSs | Distribution of securities distributed to holders of deposited securities which are distributed by the depositary to ADS holders | |
| $0.05 (or less) per ADS per calendar year | Depositary services | |
| Registration or transfer fees | Transfer and registration of ordinary shares on our share register to or from the name of the depositary or its agent when you deposit or withdraw ordinary shares | |
| Expenses of the depositary | Cable (including SWIFT) and facsimile transmissions (when expressly provided in the deposit agreement); conversion of foreign currency to U.S. dollars | |
| Taxes and other governmental charges the depositary or the custodian has to pay on any ADSs or ordinary shares underlying ADSs, such as stock transfer taxes, stamp duty, or withholding taxes | As necessary | |
| Any charges incurred by the depositary or its agents for servicing the deposited securities | As necessary |
The depositary collects its fees for delivery and surrender of ADSs directly from investors depositing shares or surrendering ADSs for the purpose of withdrawal or from intermediaries acting for them. The depositary collects fees for making distributions to investors by deducting those fees from the amounts distributed or by selling a portion of distributable property to pay the fees. The depositary may collect its annual fee for depositary services by deduction from cash distributions or by directly billing investors or by charging the book-entry system accounts of participants acting for them. The depositary may collect any of its fees by deduction from any cash distribution payable (or by selling a portion of securities or other property distributable) to ADS holders that are obligated to pay those fees. The depositary may generally refuse to provide fee-attracting services until its fees for those services are paid.
From time to time, the depositary may make payments to us to reimburse us for costs and expenses generally arising out of establishment and maintenance of the ADS program, waive fees and expenses for services provided to us by the depositary, or share revenue from the fees collected from ADS holders. In performing its duties under the deposit agreement, the depositary may use brokers, dealers, or other service providers that are affiliates of the depositary and that may earn or share fees or commissions.
144
| ITEM 13. | DEFAULTS, DIVIDEND ARREARAGES AND DELINQUENCIES |
None.
| ITEM 14. | MATERIAL MODIFICATIONS TO THE RIGHTS OF SECURITY HOLDERS AND USE OF PROCEEDS |
There are no material modifications to the rights of security holders.
| E. | Use of Proceeds |
We will not receive any proceeds from the sale of the ordinary shares represented by ADSs by the selling shareholder of ordinary shares registered pursuant to a registration statement on Form F-1, Registration Number 333-214188, which was declared effective on January 30, 2018. All net proceeds from the sale of the ordinary shares represented by ADSs will go to the selling shareholder. We may receive proceeds from the exercise of a warrant to purchase 49,607,407 ordinary shares represented by ADSs, or the Alpha Warrant, and issuance of the underlying ADSs to the extent that the Alpha Warrant is exercised for cash. The Alpha Warrant may, however, be exercisable on a cashless basis under certain circumstances. If the entire Alpha Warrant were exercised for cash in full, the proceeds would be approximately $10.3 million.
| ITEM 15. | CONTROLS AND PROCEDURES |
(a) Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer and Deputy CEO & Chief Financial Officer, has evaluated the effectiveness of our disclosure controls and procedures (as such term is defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) as of December 31, 2018, or the Evaluation Date. Based on such evaluation, those officers have concluded that, as of the Evaluation Date, our disclosure controls and procedures are effective in recording, processing, summarizing and reporting, on a timely basis, information required to be included in periodic filings under the Exchange Act and that such information is accumulated and communicated to management, including our principal executive and financial officers, as appropriate to allow timely decisions regarding required disclosure.
(b) Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over our financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the Exchange Act as a process designed by, or under the supervision of, the company’s principal executive and principal financial officers and effected by the company’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
| ● | pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transaction and dispositions of the assets of the company; |
145
| ● | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and |
| ● | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2018. In making this assessment, our management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework (2013). Based on that assessment, our management concluded that as of December 31, 2018, our internal control over financial reporting was effective.
(c) Attestation Report of the Registered Public Accounting Firm
This Annual Report on Form 20-F does not include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting due to an exemption for emerging growth companies provided in the JOBS Act.
(d) Changes in Internal Control over Financial Reporting
During the year ended December 31, 2018, there were no changes in our internal control over financial reporting that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| ITEM 16A. | AUDIT COMMITTEE FINANCIAL EXPERT |
Our board of directors has determined that each of two members of our audit committee, Mr. Scott Burell and Dr. Elan Penn, is an audit committee financial expert, as defined under the rules under the Exchange Act, and is independent in accordance with applicable Exchange Act rules and the Nasdaq Listing Rules.
| ITEM 16B. | CODE OF ETHICS |
Our board of directors has adopted a Code of Ethics applicable to all of our directors and employees, including our Chief Executive Officer, Chief Financial Officer, controller or principal accounting officer, or other persons performing similar functions, which is a “code of ethics” as defined in Item 16B of Form 20-F promulgated by the SEC. The full text of the Code of Ethics is posted on our website at www.CollPlant.com. Information contained on, or that can be accessed through, our website does not constitute a part of this a part of this Annual Report on Form 20-F and is not incorporated by reference herein. If we make any amendment to the Code of Ethics or grant any waivers, including any implicit waiver, from a provision of the Code of Ethics, we will disclose the nature of such amendment or waiver on our website to the extent required by the rules and regulations of the SEC. We have not granted any waivers under our Code of Business Conduct and Ethics.
146
| ITEM 16C. | PRINCIPAL ACCOUNTANT FEES AND SERVICES |
Kesselman & Kesselman, a member firm of PricewaterhouseCoopers International Limited, an independent registered public accounting firm, has served as our principal independent registered public accounting firm for each of the two years ended December 31, 2018 and 2017.
The following table provides information regarding fees paid by us to Kesselman & Kesselman and/or other member firms of PricewaterhouseCoopers International Limited for all services, including audit services, for the years ended December 31, 2018 and 2017:
| Year Ended December 31, |
||||||||
| 2017 | 2018 | |||||||
| (USD in thousands) | ||||||||
| Audit fees (1) | 135 | 135 | ||||||
| Tax fees(2) | 8 | 16 | ||||||
| All other fees | — | — | ||||||
| Total | 143 | 151 | ||||||
| (1) | The audit fees for the years ended December 31, 2018 and 2017 includes professional services rendered in connection with the audit of our annual consolidated financial statements and the review of our consolidated interim financial statements, statutory audits of the Company and its subsidiary, issuance of consents and assistance with review of documents filed with the SEC. |
| (2) | Tax fees for the years ended December 31, 2018 and 2017 were for services related to tax advice, including assistance with tax audit. |
Pre-Approval of Auditors’ Compensation
Our audit committee has a pre-approval policy for the engagement of our independent registered public accounting firm to perform certain audit and non-audit services. Pursuant to this policy, which is designed to assure that such engagements do not impair the independence of our auditors, the audit committee pre-approves annually a catalog of specific audit and non-audit services in the categories of audit services, audit-related services and tax services that may be performed by our independent registered public accounting firm. If a type of service, that is to be provided by our auditors, has not received such general pre-approval, it will require specific pre-approval by our audit committee. The policy prohibits retention of the independent registered public accounting firm to perform the prohibited non-audit functions defined in applicable SEC rules.
| ITEM 16D. | EXEMPTIONS FROM THE LISTING STANDARDS FOR AUDIT COMMITTEES |
Not applicable.
| ITEM 16E. | PURCHASES OF EQUITY SECURITIES BY THE ISSUER AND AFFILIATED PURCHASERS |
Not applicable.
| ITEM 16F. | CHANGE IN REGISTRANT’S CERTIFYING ACCOUNTANT |
Not applicable.
| ITEM 16G. | CORPORATE GOVERNANCE |
Under the Companies Law, companies incorporated under the laws of the State of Israel, whose shares are publicly traded, including companies whose shares are listed on the Nasdaq Capital Market are considered public companies under Israeli law and are required to comply with various corporate governance requirements under Israeli law relating to such matters as external directors, the audit committee, compensation, policy, company’s auditors, and an internal auditor. This is the case even if our shares are not listed on the Tel Aviv Stock Exchange. These requirements are in addition to the corporate governance requirements imposed by the Nasdaq Listing Rules, and other applicable provisions of U.S. securities laws to which we are subject as a foreign private issuer due to the listing of the ADSs on the Nasdaq Capital Market. Under the Nasdaq Listing Rules, a foreign private issuer, such as us, may generally follow its home country rules of corporate governance in lieu of the comparable requirements of the Nasdaq Capital Market, except for certain matters including (among others) the composition and responsibilities of the audit committee and the independence of its members within the meaning of the rules and regulations of the SEC.
147
We intend to rely on this “home country practice exemption” with respect to the following Nasdaq Listing Rules:
| ● | Quorum requirements. As permitted under the Companies Law pursuant to our articles of association, the quorum required for an ordinary meeting of shareholders will consist of at least two shareholders present in person, by proxy or by other voting instrument in accordance with the Companies Law, who hold at least 20% of the voting power of our shares (and in an adjourned meeting, with some exceptions, any number of participating shareholders), instead of 331/3% of the issued share capital required under the Nasdaq Listing Rules. |
| ● | Distribution of certain reports to shareholders. As opposed to the Nasdaq Listing Rules, which require listed issuers to make its annual reports available to shareholders in one of a number of specific manners, Israeli law does not require that we distribute annual reports, including our financial statements. As such, the generally accepted business practice in Israel is to distribute such reports to shareholders through a public regulated distribution website. In addition to making such reports available on a public regulated distribution website, we plan to make our audited financial statements available to our shareholders at our offices and will only mail such reports to shareholders upon request. As a foreign private issuer, we are generally exempt from the SEC’s proxy solicitation rules. |
| ● | Shareholder approval. We will seek shareholder approval for all corporate actions requiring such approval under the requirements of the Companies Law, rather than seeking approval for corporate actions in accordance with Nasdaq Listing Rule 5635. In particular, under this Nasdaq Listing Rule, shareholder approval is generally required for: (i) an acquisition of shares or assets of another company that involves the issuance of 20% or more of the acquirer’s shares or voting rights or if a director, officer or 5% shareholder has greater than a 5% interest in the target company or the consideration to be received; (ii) the issuance of shares leading to a change of control; (iii) adoption or amendment of equity compensation arrangements; and (iv) issuances of 20% or more of the shares or voting rights (including securities convertible into, or exercisable for, equity) of a listed company via a private placement (or via sales by directors, officers or 5% shareholders) if such equity is issued (or sold) at below the greater of the book or market value of shares. By contrast, under the Companies Law, shareholder approval is required (subject to certain limited exceptions) for, among other things: (a) transactions with directors concerning the terms of their service (including indemnification, exemption, and insurance for their service or for any other position that they may hold at a company), for which approvals of the compensation committee, board of directors, and shareholders are all required; (b) extraordinary transactions with controlling shareholders of publicly held companies, which require the special approval described below under “Disclosure of Personal Interests of Controlling Shareholders and Approval of Certain Transactions;” (c) terms of office and employment or other engagement of our controlling shareholder, if any, or such controlling shareholder’s relative, which require the special approval described below under “Disclosure of Personal Interests of Controlling Shareholders and Approval of Certain Transactions;” (d) approval of transactions with Company’s Chief Executive Officer with respect to his or hers compensation, whether in accordance with the approved compensation policy of the Company or not in accordance with the approved compensation policy of the Company, or transactions with officers of the Company not in accordance with the approved compensation policy; and (e) approval of the compensation policy of the Company for office holders. In addition, under the Companies Law, a merger requires approval of the shareholders of each of the merging companies. |
Except as stated above, we intend to comply with the rules generally applicable to U.S. domestic companies listed on the Nasdaq Capital Market, subject to certain exemptions the JOBS Act provides to emerging growth companies. We may in the future decide to use other foreign private issuer exemptions with respect to some or all of the other Nasdaq Listing Rules. Following our home country governance practices, as opposed to the requirements that would otherwise apply to a company listed on the Nasdaq Capital Market, may provide less protection than is accorded to investors under the Nasdaq Listing Rules applicable to domestic issuers.
| ITEM 16H. | MINE SAFETY DISCLOSURE |
Not applicable.
148
| ITEM 17. | FINANCIAL STATEMENTS |
We have elected to provide financial statements and related information pursuant to Item 18.
| ITEM 18. | FINANCIAL STATEMENTS |
The consolidated financial statements and the related notes required by this Item are included in this Annual Report on Form 20-F beginning on page F-1.
| ITEM 19. | EXHIBITS. |
149
150
| * | Filed herewith. |
| † | Portions of this exhibit have been omitted and filed separately with the Securities and Exchange Commission pursuant to a confidential treatment request. |
| # | Management contract or compensatory plan. |
151
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this Annual Report on Form 20-F filed on its behalf.
| COLLPLANT HOLDINGS LTD. | ||
| Date: April 1, 2019 | By: | /s/ Eran Rotem |
| Eran Rotem | ||
| Deputy CEO and Chief Financial Officer | ||
152

REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the board of directors and shareholders of CollPlant Holdings Ltd.
Opinion on the Financial Statements
We have audited the accompanying consolidated statements of financial position of CollPlant Holdings Ltd. and its subsidiary (the “Company”) as of December 31, 2018 and 2017, and the related consolidated statements of comprehensive loss, changes in equity and cash flows for each of the three years in the period ended December 31, 2018, including the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2018 and 2017, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2018 in conformity with International Financial Reporting Standards as issued by the International Accounting Standards Board.
Substantial Doubt About the Company’s Ability to Continue as a Going Concern
The accompanying consolidated financial statements have been prepared assuming the Company will continue as a going concern. As discussed in Note 1a to the consolidated financial statements, the Company has suffered recurring losses from operations and cash outflows from operating activities that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1a. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits of these consolidated financial statements in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
| Tel-Aviv, Israel | /s/ Kesselman & Kesselman | |
| March 31, 2019 | Certified Public Accountants (lsr.) | |
| A member firm of PricewaterhouseCoopers International Limited |
We have served as the Company’s auditor since 2005.
F-1
CollPlant Holdings Ltd.
Consolidated Statements of Financial Position
| December 31, | Convenience translation into USD (Note 1b) December 31, |
|||||||||||||
| Note | 2017 | 2018 | 2018 | |||||||||||
| NIS in thousands | In thousands | |||||||||||||
| Assets | ||||||||||||||
| Current assets: | ||||||||||||||
| Cash and cash equivalents | 5 | 17,817 | 20,065 | 5,354 | ||||||||||
| Accounts receivables: | ||||||||||||||
| Trade receivables | 354 | 1,933 | 516 | |||||||||||
| Other | 6 | 3,543 | 1,253 | 334 | ||||||||||
| Restricted deposit | - | 584 | 154 | |||||||||||
| Inventory | 2(I) | 700 | 3,052 | 814 | ||||||||||
| 22,414 | 26,887 | 7,172 | ||||||||||||
| Non-current assets: | ||||||||||||||
| Restricted deposit | 503 | 580 | 155 | |||||||||||
| Long term-receivables | 92 | 68 | 18 | |||||||||||
| Property and equipment, net | 7 | 3,582 | 5,274 | 1,407 | ||||||||||
| Intangible assets, net | 8 | 1,454 | 1,273 | 340 | ||||||||||
| 5,631 | 7,195 | 1,920 | ||||||||||||
| Total assets | 28,045 | 34,082 | 9,092 | |||||||||||
| Liabilities and equity | ||||||||||||||
| Current liabilities: | ||||||||||||||
| Loan | - | 84 | 22 | |||||||||||
| Accounts payable: | 10 | |||||||||||||
| Trade payables | 2,922 | 2,331 | 622 | |||||||||||
| Accrued liabilities and other | 1,996 | 2,366 | 631 | |||||||||||
| Contract liabilities | 2(N),13C | - | 3,636 | 970 | ||||||||||
| 4,918 | 8,417 | 2,245 | ||||||||||||
| Non-current liabilities: | ||||||||||||||
| Debentures at fair value | 4,12 | 12,639 | ||||||||||||
| Warrants at fair value | 4,12 | 2,434 | 649 | |||||||||||
| Derivatives | 4,12 | 141 | 363 | 97 | ||||||||||
| Royalties to the Israel Innovation Authority | 13(A)(2) | 1,203 | 1,185 | 316 | ||||||||||
| Loan | - | 84 | 22 | |||||||||||
| Long term contract liabilities | - | 3,673 | 980 | |||||||||||
| Long-term payables | 61 | - | - | |||||||||||
| 14,044 | 7,739 | 2,064 | ||||||||||||
| Commitments and contingent liabilities | 13 | |||||||||||||
| Total liabilities | 18,962 | 16,156 | 4,309 | |||||||||||
| Equity: | 14 | |||||||||||||
| Ordinary shares | 4,998 | 5,716 | 1,525 | |||||||||||
| Additional paid in capital and warrants | 178,467 | 196,870 | 52,527 | |||||||||||
| Accumulated deficit | (174,382 | ) | (184,660 | ) | (49,269 | ) | ||||||||
| Total equity | 9,083 | 17,926 | 4,783 | |||||||||||
| Total liabilities and equity | 28,045 | 34,082 | 9,092 | |||||||||||
The accompanying notes are an integral part of the consolidated financial statements
F-2
CollPlant Holdings Ltd.
Consolidated Statements of Comprehensive Loss
| Year ended December 31, | Convenience translation into USD (Note 1b) |
|||||||||||||||||
| Note | 2016 | 2017 | 2018 | 2018 | ||||||||||||||
| NIS in thousands | In thousands | |||||||||||||||||
| Revenue | 15 | 292 | 1,668 | 18,034 | 4,812 | |||||||||||||
| Cost of revenue | - | 52 | 1,426 | 380 | ||||||||||||||
| Gross profit | 292 | 1,616 | 16,608 | 4,432 | ||||||||||||||
| Research and development expenses: | 16 | |||||||||||||||||
| Research and development expenses | 29,200 | 16,921 | 16,213 | 4,325 | ||||||||||||||
| Participation in research and development expenses, net | (12,411 | ) | (2,855 | ) | 3,227 | 861 | ||||||||||||
| Research and development expenses, net | 16,789 | 14,066 | 19,440 | 5,186 | ||||||||||||||
| General, administrative and marketing expenses | 17 | 11,048 | 8,303 | 12,480 | 3,330 | |||||||||||||
| Operating loss | (27,545 | ) | (20,753 | ) | (15,312 | ) | (4,084 | ) | ||||||||||
| Financial income | 18 | 93 | 253 | 1,650 | 440 | |||||||||||||
| Financial expenses | 18 | (441 | ) | (380 | ) | (225 | ) | (60 | ) | |||||||||
| Financial income (expenses), net | (348 | ) | (127 | ) | 1,425 | 380 | ||||||||||||
| Comprehensive loss | 27,893 | 20,880 | 13,887 | 3,704 | ||||||||||||||
| Basic and diluted loss per ordinary share (NIS/USD) | 19 | 0.28 | 0.16 | 0.06 | 0.02 | |||||||||||||
| Weighted average ordinary shares outstanding | 100,624,945 | 133,187,048 | 219,229,229 | 219,229,229 | ||||||||||||||
The accompanying notes are an integral part of the consolidated financial statements.
F-3
CollPlant Holdings Ltd.
Consolidated Statements of Changes in Equity
| Ordinary shares |
Additional paid in capital and warrants |
Accumulated deficit |
Total equity | |||||||||||||
| NIS in thousands | ||||||||||||||||
| Balance as at January 1, 2016 | 2,665 | 140,704 | (133,590 | ) | 9,779 | |||||||||||
| Movement in 2016: | ||||||||||||||||
| Comprehensive loss for the year | - | - | (27,893 | ) | (27,893 | ) | ||||||||||
| Share-based compensation to employees and consultants | - | - | 3,572 | 3,572 | ||||||||||||
| Proceeds from issuance of shares and options, less issuance expenses of NIS 1,327 thousand | 510 | 17,995 | - | 18,505 | ||||||||||||
| Issuance of shares, See Note 13(A)(1)(D) | 32 | 1,165 | - | 1,197 | ||||||||||||
| Balance as at December 31, 2016 | 3,207 | 159,864 | (157,911 | ) | 5,160 | |||||||||||
| Movement in 2017: | ||||||||||||||||
| Comprehensive loss for the year | - | - | (20,880 | ) | (20,880 | ) | ||||||||||
| Share-based compensation to employees and consultants | 32 | 529 | 4,409 | 4,970 | ||||||||||||
| Proceeds from issuance of shares and warrants, net of issuance expenses of NIS 634 thousand | 1,457 | 14,758 | - | 16,215 | ||||||||||||
| Exercise of warrants into shares | 302 | 3,316 | - | 3,618 | ||||||||||||
| Balance as at December 31, 2017 | 4,998 | 178,467 | (174,382 | ) | 9,083 | |||||||||||
| Movement in 2018: | ||||||||||||||||
| Comprehensive loss for the year | - | - | (13,887 | ) | (13,887 | ) | ||||||||||
| Share-based compensation to employees and consultants | 6 | 24 | 3,609 | 3,639 | ||||||||||||
| Conversion of Debentures to Prepaid warrants | 12,708 | - | 12,708 | |||||||||||||
| Conversion of Prepaid warrants to Ordinary shares | 248 | (248 | ) | - | - | |||||||||||
| Proceeds from issuance of shares, net of issuance expenses of NIS 350 thousand | 464 | 5,919 | - | 6,383 | ||||||||||||
| Balance as at December 31, 2018 | 5,716 | 196,870 | (184,660 | ) | 17,926 | |||||||||||
| Convenience translation into
USD (Note 1b) in thousands |
||||||||||||||||
| Balance as at January 1, 2018 | 1,333 | 47,617 | (46,527 | ) | 2,423 | |||||||||||
| Movement in 2018: | ||||||||||||||||
| Comprehensive loss for the year | - | - | (3,704 | ) | (3,704 | ) | ||||||||||
| Share-based compensation to employees and consultants | 2 | 6 | 962 | 970 | ||||||||||||
| Conversion of Debentures to Prepaid warrants | 3,391 | - | 3,391 | |||||||||||||
| Conversion of Prepaid warrants to Ordinary shares | 66 | (66 | ) | - | ||||||||||||
| Proceeds from issuance of shares, net of issuance expenses of $ 93 thousand | 124 | 1,579 | - | 1,703 | ||||||||||||
| Balance as at December 31, 2018 | 1,525 | 52,527 | (49,269 | ) | 4,783 | |||||||||||
The accompanying notes are an integral part of the consolidated financial statements
F-4
CollPlant Holdings Ltd.
Consolidated Statements of Cash Flows
| Year ended December 31, | Convenience translation into USD (Note 1b) |
|||||||||||||||
| 2016 | 2017 | 2018 | 2018 | |||||||||||||
| NIS in thousands | In thousands | |||||||||||||||
| Cash flows from operating activities: | ||||||||||||||||
| Net cash used in operations (see Appendix A) | (19,357 | ) | (17,884 | ) | (4,665 | ) | (1,245 | ) | ||||||||
| Net cash used in operating activities | (19,357 | ) | (17,884 | ) | (4,665 | ) | (1,245 | ) | ||||||||
| Cash flows from investing activities: | ||||||||||||||||
| Restricted deposits | - | - | (620 | ) | (166 | ) | ||||||||||
| Purchase of property and equipment | (492 | ) | (447 | ) | (2,983 | ) | (796 | ) | ||||||||
| Net cash used in investing activities | (492 | ) | (447 | ) | (3,603 | ) | (962 | ) | ||||||||
| Cash flows from financing activities: | ||||||||||||||||
| Proceeds from issuance of shares, warrants and debentures, less issuance expenses | 18,505 | 29,030 | 9,999 | 2,668 | ||||||||||||
| Exercise of options and warrants into shares | - | 3,618 | - | - | ||||||||||||
| Loan received | - | - | 210 | 57 | ||||||||||||
| Loan paid | - | - | (42 | ) | (11 | ) | ||||||||||
| Payments made for equipment on financing terms | (19 | ) | (253 | ) | (252 | ) | (67 | ) | ||||||||
| Net cash provided by financing activities | 18,486 | 32,395 | 9,915 | 2,647 | ||||||||||||
| Increase (Decrease) in cash and cash equivalents | (1,363 | ) | 14,064 | 1,647 | 440 | |||||||||||
| Cash and cash equivalents at the beginning of the year | 5,317 | 3,797 | 17,817 | 4,754 | ||||||||||||
| Exchange differences on cash and cash equivalents | (157 | ) | (44 | ) | 601 | 160 | ||||||||||
| Cash and cash equivalents at the end of the year | 3,797 | 17,817 | 20,065 | 5,354 | ||||||||||||
The accompanying notes are an integral part of the consolidated financial statements
F-5
CollPlant Holdings Ltd.
Appendices to the Consolidated Statements of Cash Flows
| Year ended December 31, | Convenience translation into USD (Note 1b) |
|||||||||||||||
| 2016 | 2017 | 2018 | 2018 | |||||||||||||
| NIS in thousands | In thousands | |||||||||||||||
| Appendix to the statement of cash flows | ||||||||||||||||
| A. Net cash used in operations: | ||||||||||||||||
| Comprehensive loss for the year | (27,893 | ) | (20,880 | ) | (13,887 | ) | (3,704 | ) | ||||||||
| Adjustments for: | ||||||||||||||||
| Depreciation and amortization | 864 | 1,050 | 1,472 | 392 | ||||||||||||
| Share-based compensation to employees and consultants | 3,572 | 4,970 | 5,169 | 1,379 | ||||||||||||
| Exchange differences on cash and cash equivalents | 157 | 44 | (601 | ) | (160 | ) | ||||||||||
| Changes in fair value of financial instruments | - | (35 | ) | (891 | ) | (238 | ) | |||||||||
| Exchange differences on restricted deposit | 8 | 54 | (41 | ) | (11 | ) | ||||||||||
| (23,292 | ) | (14,797 | ) | (8,779 | ) | (2,342 | ) | |||||||||
| Changes in operating asset and liability items: | ||||||||||||||||
| Increase in trade receivables | (544 | ) | (137 | ) | (1,579 | ) | (421 | ) | ||||||||
| Increase in inventory | (487 | ) | (213 | ) | (2,352 | ) | (628 | ) | ||||||||
| Decrease (increase) in other receivables (including long-term receivables) | (95 | ) | 101 | 784 | 209 | |||||||||||
| Increase (decrease) in trade payables (including long-term payables) | 2,498 | (2,239 | ) | (400 | ) | (107 | ) | |||||||||
| Increase in accrued liabilities and other payables | 382 | 379 | 370 | 99 | ||||||||||||
| Increase in contract liabilities (including long term contract liabilities) | - | - | 7,309 | 1,950 | ||||||||||||
| Increase (decrease) in royalties to the IIA | 2,181 | (978 | ) | (18 | ) | (5 | ) | |||||||||
| 3,935 | (3,087 | ) | 4,114 | 1,097 | ||||||||||||
| Net cash used in operations | (19,357 | ) | (17,884 | ) | (4,665 | ) | (1,245 | ) | ||||||||
| B. Non-cash investing and financing activities | ||||||||||||||||
| Acquisition of fixed assets against issuance of shares and credit See Note 13(A)(1)(C). | 1,678 | - | - | - | ||||||||||||
| Conversion of Debentures to pre-paid warrant | - | - | 12,708 | 3,391 | ||||||||||||
| Conversion of pre-paid warrants to ordinary shares | - | - | 248 | 66 | ||||||||||||
The accompanying notes are an integral part of the consolidated financial statements
F-6
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 1—GENERAL
| A. | CollPlant Holdings Ltd. is a regenerative medicine company focused on developing and commercializing tissue repair products, initially for three-dimensional bio-printing of tissues and organs, dermal fillers for, aesthetics, orthobiologics and advanced wound care markets. CollPlant’s products are based on its rhCollagen, a form of human collagen produced with CollPlant’s proprietary plant-based genetic engineering technology. The Company sales includes sales of (i) the BioInk product for the development of 3D bioprinting of organs and tissues and (ii) sales in Europe of the products for tendinopathy and wound healing, that received during 2016 a CE approval that enables their marketing in Europe. |
The Company operates through CollPlant Ltd., a wholly-owned subsidiary (CollPlant Holdings Limited and CollPlant Ltd. will be referred to hereinafter as “the Company” and “CollPlant”, respectively).
The address of the Company’s registered office is 4 Oppenheimer St., Science Park, Rehovot, Israel. Since January 31, 2018, the Company’s American Depositary Shares (“ADSs”) commenced trading on the Nasdaq Capital Market. Each ADS represents 50 ordinary shares. On October 29, 2018 the Company’s ordinary shares delisted from the Tel Aviv Stock Exchange (“TASE”).
The Company has an accumulated deficit as of December 31, 2018, as well as a history of net losses and negative operating cash flows in recent years. The Company has an accumulated deficit of approximately NIS 184.7 million as of December 31, 2018. The Company expects to continue incurring losses and negative cash flows from operations until its products (primarily BioInk) reach commercial profitability. As a result of these expected losses and negative cash flows from operations, along with the Company’s current cash position, the Company does not have sufficient cash to meet its liquidity requirements for the following twelve months. Consequently, there is substantial doubt about the Company’s ability to continue as a going concern. These financial statements have been prepared assuming that the Company will continue as a going concern and do not include any adjustments that might result from the outcome of this uncertainty.
Management’s plans include the continued commercialization of the Company’s products and raising capital through the sale of additional equity securities, debt or capital inflows from strategic partnerships. There are no assurances however, that the Company will be successful in obtaining the level of financing needed for its operations. If the Company is unsuccessful in commercializing its products and raising capital, it may need to reduce activities, curtail or cease operations.
| B. | Convenience translation into U.S. dollars (“dollars”, “USD” or “$”) |
For the convenience of the reader, the reported New Israeli Shekel (“NIS”) amounts as of December 31, 2018 and for the year then ended have been translated into U.S. dollars at the Bank of Israel’s representative rate of exchange for December 31, 2018 ($1 = NIS 3.748). The dollar amounts presented in these financial statements should not be construed as representing amounts that are receivable or payable in dollars or convertible into dollars, unless otherwise indicated.
| C. | Approval of financial statements |
These consolidated financial statements were approved by the board of directors on March 31, 2019.
F-7
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 2—SIGNIFICANT ACCOUNTING POLICIES
| A. | Basis of presentation of the financial statements |
The Company’s financial statements as of December 31, 2017 and 2018 and for each of the three years ended on December 31, 2018 comply with International Financial Reporting Standards (“IFRS”) and interpretations issued by the IFRS Interpretations Committee (“IFRS IC”) applicable to companies reporting under IFRS, as issued by the International Accounting Standard Board (“IASB”).
The significant accounting policies described below have been applied consistently to all the years presented, unless otherwise stated.
The consolidated financial statements have been prepared on the basis of historical cost except for debentures and derivatives at fair value.
The preparation of financial statements that comply with IFRS requires the use of certain critical accounting estimates. It also requires management to exercise judgment when applying the Company’s accounting policies. Note 3 provides disclosure of areas involving a considerable degree of judgment or complexity, or areas where assumptions and estimates have a material effect on the financial statements. Actual results may differ materially from the estimates and assumptions used by the Company’s management.
| B. | Consolidated financial statements |
A subsidiary is an entity over which the Company has control. The Company controls an entity when the Company is exposed to, or has rights to, variable returns from its involvement with the entity and has the ability to affect those returns through its power over the entity. Subsidiaries are fully consolidated from the date on which control is transferred to the Company. The subsidiaries are deconsolidated from the date that control ceases.
| C. | Translation of foreign currency balances and transactions: |
| 1) | Functional currency and presentation currency |
Items included in the financial statements are measured using the currency of the primary economic environment in which the Company operates (“Functional Currency”). The financial statements are stated in NIS, which is the Functional Currency and presentation currency of the Company and its subsidiary.
| 2) | Transactions and balances |
Transactions in currencies other than the functional currency (“Foreign Currencies”) are translated into the Functional Currency at exchange rates at the dates of transaction. Foreign exchange gains and losses resulting from the settlement of such transactions and from the translation at year-end exchange rates of monetary assets and liabilities denominated in Foreign Currencies are recognized in the profit or loss for the year.
Gains and losses arising from changes in exchange rates are recognized in the statement of comprehensive loss under “Financing expenses (income), net”.
F-8
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 2—SIGNIFICANT ACCOUNTING POLICIES (continued)
| D. | Property and equipment |
| 1) | All property and equipment (including leasehold improvements) are stated at historical cost less accumulated depreciation and impairment. Historical cost of an item of property and equipment includes: |
| a. | Its purchase price, including import duties and non-refundable purchase taxes, after deducting trade discount and rebates. |
| b. | Any costs directly attributable to bringing the asset to the location and condition necessary for it to be capable of operating in the manner intended by management. |
Repairs and maintenance are charged to the statements of comprehensive loss during the period in which they are incurred.
| 2) | The assets are depreciated using the straight-line method to allocate their cost over their estimated useful lives, as follows: |
| Years | ||
| Computer equipment | 3 | |
| Greenhouse equipment* | 4 - 10 | |
| Office furniture | 7 - 17 | |
| Laboratory equipment | 4 - 5 | |
| Leasehold improvements | 5 | |
| Vehicles | 4-7 |
| * | Greenhouse equipment—agricultural equipment used in the tobacco production greenhouse. |
Leasehold improvements are depreciated over the lease period or the expected useful life of the improvements, whichever is shorter.
Impairment of the asset to its recoverable amount is recognized as incurred, if the carrying amount of the asset is greater than its estimated recoverable amount (see also section F below).
| 3) | Gains or losses on disposals are determined by comparing net proceeds with the carrying amount. These are included in the statement of comprehensive loss. |
| E. | Intangible assets |
| 1) | In process research and development (“IPR&D”) |
Acquired IPR&D is presented based on the fair value at the date of the acquisition and up to December 31, 2015 (see below), was not depreciated. Such asset was tested annually for impairment, see section F below. The assessment was carried out more frequently if there were indications of impairment.
F-9
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 2—SIGNIFICANT ACCOUNTING POLICIES (continued)
Up to December 31, 2015, during the research and development period, this intangible asset was not amortized. Commencing 2016, the said asset is available for use and therefore is amortized on a straight-line basis until the end of the period of the patent for the know-how (approximately 10 years).
For information about impairment of non-monetary assets, see F below.
| 2) | Software |
Acquired software licenses are capitalized on the basis of the cost incurred to acquire and implement the specific software. These costs are amortized on a straight-line basis over the estimated useful life of licenses (3 years).
| 3) | Research and development (“R&D”) |
Research expenses are recognized as an expense as incurred. Costs incurred for development projects (referring to design and testing of new or improved products) are recognized as intangible assets when the following conditions exist:
| ● | It is technically feasible to complete the intangible asset so that it will be available for use; |
| ● | Management intends to complete the development of the intangible asset and to use or sell the asset; |
| ● | The intangible asset can be used or sold; |
| ● | It can be demonstrated how the intangible asset will generate probable future economic benefits; |
| ● | There are adequate technical, financial and other resources to complete development and to use or sell the intangible asset; |
| ● | The expenditure attributable to the intangible asset can be reliably measured during its development. |
Other development costs that do not meet these criteria are recognized as an expense when incurred. Development costs previously recognized as an expense are not recognized as an asset in subsequent periods.
As of December 31, 2018, the Company has not met the rules for capitalizing development costs as an intangible asset and accordingly, no asset whatsoever has been recognized in the financial statements for such costs.
F-10
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 2—SIGNIFICANT ACCOUNTING POLICIES (continued)
| F. | Impairment of non-monetary assets |
Assets that have indefinite useful life are not subject to amortization and are tested annually (or when there are indicators for impairment-see below) for impairment.
All non-monetary assets are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount may not be recoverable. An impairment loss is recognized for the amount by which the asset’s carrying amount exceeds its recoverable amount. The recoverable amount of an asset is the higher of its fair value less costs to sell and value in use. For the purpose of assessing impairment, assets are grouped together at the lowest levels for which there are separately identifiable cash flows (cash-generating units). Non-financial assets other than goodwill that suffered impairment are reviewed for possible reversal of the impairment at each reporting date.
For the years ended December 31, 2016, 2017 and 2018, no impairment has been recognized.
| G. | Government grants |
Government grants, which are received from the Israel Innovation Authority (“IIA”) (formerly known as the Israeli Office of Chief Scientist or OCS) by way of participation in research and development that is conducted by the Company, fall within the scope of “forgivable loans,” as set forth in International Accounting Standard 20 “Accounting for Government Grants and Disclosure of Government Assistance” (“IAS 20”).
As approved by the IIA, the grants are received in installments as the program progresses. The Company recognizes each forgivable loan on a systematic basis at the same time the Company records, as an expense, the related research and development costs for which the grant is received, provided that there is reasonable assurance that: (a) the Company complies with the conditions attached to the grant, and (b) the grant will be received (usually upon receipt of approval notice). The amount of the forgivable loan is recognized based on the participation rate approved by the IIA; thus, a forgivable loan is recognized as a receivable when approved research and development costs have been incurred before grant funds are received.
If at the time of grant approval there is reasonable assurance that the Company will comply with the forgivable loan conditions attached to the grant, and that the Company will not pay royalties to IIA, grant income is recorded against the related research and development expenses in the statements of comprehensive loss.
If at the time of grant or in subsequent periods, it is not reasonably assured that royalties will not be paid to the IIA, the Company recognizes a liability that is measured based on the Company’s best estimate of the amount required to settle the Company’s obligation at the end of each reporting period.
F-11
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 2—SIGNIFICANT ACCOUNTING POLICIES (continued)
| H. | Cash and cash equivalents |
Cash and cash equivalents include cash on hand and short-term bank deposits, and other short-term highly liquid investments with original maturities of three months or less.
| I. | Inventory |
Inventory is measured at the lower of cost and net realizable value.
The cost of inventories is based on the first-in first-out (FIFO) principle. In the case of purchased goods and work in process, costs include design, raw materials, direct labor, other direct costs and fixed production overheads (based on the normal operating capacity of the production facilities).
Net realizable value is the estimated selling price in the ordinary course of business, less variable attributable selling expenses.
| J. | Share capital and other equity instruments |
The Company’s ordinary shares are classified as share capital. Incremental costs directly attributable to the issue of new shares or warrants are recognized in equity as a deduction of issue proceeds. Pre-paid warrants (that their exercise price was prepaid) that upon exercise will be converted into fixed amount of ordinary shares – are classified as equity instruments in the statements of financial position.
| K. | Trade payables |
Trade payables include the Company’s liabilities to pay for goods or services purchased from suppliers in the ordinary course of business. Trade payables are classified as current liabilities if payment is due within one year, otherwise they are recognized as non-current liabilities.
Trade payables are recognized initially at fair value and subsequently measured at amortized cost based on the effective interest method.
F-12
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 2—SIGNIFICANT ACCOUNTING POLICIES (continued)
| L. | Deferred taxes |
The Company recognizes deferred taxes based on the liability method, for temporary differences between the carrying amounts of assets and liabilities included in the consolidated financial statements and the amounts used for tax purposes. However, deferred tax liabilities are not recognized if they arise from the initial recognition of goodwill. In addition, deferred taxes are not recognized if the temporary differences arise on initial recognition of an asset or a liability, other than in a business combination, which, at the time of the transaction, have no effect on profit or loss—whether for accounting or tax purposes. The amount of deferred taxes is determined in accordance with the tax rates (and tax laws) that have been enacted or substantively enacted as at the date of the statement of financial position and are expected to apply when the deferred tax assets will be realized or when the deferred tax liabilities will be settled.
Deferred tax assets are recognized for deductible temporary differences, to the extent that it is probable that future taxable profits will be available against which they can be utilized.
In the absence of a forecast of future taxable income, a deferred tax asset was not recognized in the Company’s financial statements.
| M. | Employee benefits |
| 1) | Liability for severance pay |
In accordance with labor laws and labor agreements in effect, the Company and its subsidiary are required to pay severance and pension benefits to employees who are dismissed or retire under certain circumstances.
The said liability to pay pension and severance pay is related to employees in Israel who are covered by Section 14 of the Severance Pay Law, and is covered by regular contributions to defined contribution plans. The amounts contributed are not included in the statement of financial position.
| 2) | Vacation and recreation pay |
By law, all employees are entitled to vacation and recreation pay, calculated on a monthly basis. The right is based on the employment period.
F-13
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 2—SIGNIFICANT ACCOUNTING POLICIES (continued)
| N. | Revenue recognition |
General
As of January 1, 2018, the Company has applied IFRS 15 using the modified retrospective approach, in accordance with the transitional directive, which allows recognition of the cumulative effect of the initial application as an adjustment to the opening balance of equity of initial application.
The initial implementation of IFRS 15 did not have a material effect on the consolidated financial statements of the Company.
IFRS 15 introduces a five-step model for recognizing revenue from contracts with customers, as follows:
| (1) | Identifying the contract(s) with the customer. |
| (2) | Identifying the separate performance obligations in the contract. |
| (3) | Determining the transaction price. |
| (4) | Allocating the transaction price to separate performance obligations in the contract. |
| (5) | Recognizing revenue when (or as) each of the performance obligations is satisfied. |
Revenue from the sale of goods
Revenue from sale of goods is recognized in profit or loss at the point in time when the control of the goods is transferred to the customer, generally upon delivery of the goods to the customer. The goods are products based on the Company’s rhCollagen, and includes the BioInk product for the development of 3D bioprinting of organs and tissues and products for tendinopathy and wound healing.
Revenue from rendering of services
Revenue from rendering of services is recognized over time, during the period the customer simultaneously receives and consumes the benefits provided by the Company’s performance. Under the Company’s service contracts, the Company has a right to consideration from the customer in an amount that corresponds directly with the value to the customer of the Company’s performance completed to date. Therefore, the Company utilizes the practical expedient in IFRS 15 and recognizes revenue in the amount to which the Company has a right to invoice.
The Company charges its customers based on payment terms agreed upon in specific agreements. When payments are made before or after the service is performed, the Company recognizes the resulting contract asset or liability.
F-14
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 2—SIGNIFICANT ACCOUNTING POLICIES (continued)
Revenues from licensing agreement
On October 19, 2018, the Company signed a License, Development and Commercialization Agreement (the “License Agreement”) with Lung Biotechnology PBC (“LB”), a public benefit corporation and wholly-owned subsidiary of United Therapeutics Corporation, pursuant to which LB will be entitled to develop engineered lungs or lung substitutes using CollPlant’s rhCollagen and BioInk (see also Note 13(c)).
According to IFRS 15, a performance obligation is a promise to provide a distinct good or service or a series of distinct goods or services. Goods and services that are not distinct are bundled with other goods or services in the contract until a bundle of goods or services that is distinct is created. A good or service promised to a customer is distinct if the customer can benefit from the good or service either on its own or together with other resources that are readily available to the customer and the entity’s promise to transfer the good or service to the customer is separately identifiable from other promises in the contract.
Options granted to the customer that do not provide a material right to the customer that it would not receive without entering into the contract do not give rise to performance obligations.
The Company has identified the following performance obligations in the License Agreement: (1) grant of the license and use of its IP (“License”); and (2) a limited quantity of BioInk to be supplied over a specific time frame (“First BioInk”). The License is distinct as the licensee is able to benefit from the license on its own at its current stage (inter alia, due to sublicensing rights, option services can be obtained from other experts in the field and not necessarily from the Company, etc.).
F-15
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 2—SIGNIFICANT ACCOUNTING POLICIES (continued)
In addition, the Company has identified several options in the License Agreement. However, neither of the options provides a material right to the customer and therefore, neither of the said options give rise to a performance obligation. The said options are: (1) grant of two years option to extend the License to additional organs or organ substitutes (“Products Option”); (2) one-year right of first refusal to receive an exclusive license relating to the option products (“Right of First Refusal”); (3) option for the purchase of additional BioInk that may be supplied (“Additional BioInk”); and (4) option for the purchase of consulting services (“Consulting Services”).
The transaction price included an up-front paid amount of $5.0 million and reimbursement for part of the costs related to the IIA in an amount of $1.0 million, as well as variable considerations contingent upon LB achieving certain milestones, sales-based royalties and additional reimbursement of costs related to payments made by CollPlant to the IIA (“Variable Consideration”).
IFRS 15 defines the ‘Transaction Price’ as the amount of consideration to which the entity expects to be entitled in exchange for transferring the promised goods or services to a customer. The Company allocates the transaction price to each performance obligation identified based on the standalone selling prices of the goods or services being provided to the customer. The stand-alone selling price is the price at which the Company would sell the promised goods or services separately to a customer. When the stand-alone selling price is not directly observable by reference to similar transactions with similar customers, the Company applies suitable methods for estimating the stand-alone selling price.
In addition, as stated above, Variable Consideration is included in the transaction price at its “most likely outcome” and only to the extent that it is highly probable that a significant reversal in the amount of revenue recognized will not occur when the uncertainty associated with the Variable Consideration is subsequently resolved.
With respect to the Variable Consideration:
| a) | Sales-based royalties are not included in the transaction price. Rather, they are recognized as incurred, due to the specific exception of IFRS 15 for sales-based royalties in licensing of intellectual properties. | |
| b) | The Variable Consideration is related to LB achieving certain milestones, including the related reimbursement of part of the IIA royalties. Since the Company estimates that the “most likely outcome” of these milestones would not be achieved, it is not included in the transaction price. The said Variable Consideration will be recognized only when the “most likely outcome” would be that milestones will be achieved and it would be highly probable that a significant reversal of revenues will not occur, usually only upon achievement of the specific milestone. |
The following are the details of the allocation of the transaction price (which does not include the Variable Consideration) to the various performance obligations in the Agreement:
| a) | The First BioInk was allocated with its stand-alone selling price, which is the observable price of the BioInk when the Company sells it separately. | |
| b) | The License was allocated with an estimated stand-alone selling price, based on the residual approach, since the Company has not yet established a price for that license and the license has not previously been sold on a stand-alone basis (i.e. the selling price is uncertain), as well as the related IIA royalties reimbursement. |
F-16
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 2—SIGNIFICANT ACCOUNTING POLICIES (continued)
The License performance obligation is considered a right to use IP in accordance with IFRS 15. Therefore, the transaction price allocated to it was recognized at a point in time, upon transfer of control over the License to LB.
The transaction price allocated to the First BioInk is recognized at the point in time when the control of the BioInk is transferred to LB, generally upon delivery of the BioInk to LB.
The Company considered whether the transaction price allocated to the First BioInk includes a financing component and concluded that the financing component is not material.
| O. | Share-based payment |
The Company has a share-based payment plan for employees and service providers, settled by the Company’s equity instruments, whereby the Company receives services from employees and service providers in exchange for the Company’s equity instruments (options or shares). The fair value of services received from employees and service providers in exchange for the equity instruments is recognized as an expense in the statements of comprehensive loss. With respect to options granted to employees the total amount recognized as an expense in statements of the comprehensive loss is based on the fair value of the options granted, without taking into account the effect of service conditions and non-market vesting conditions.
With respect to options granted to service providers and suppliers, the fair value of the grant is determined in accordance with the fair value of the service or goods received.
Non-market vesting conditions are included in the assumptions used to estimate the number of options expected to vest. The total expense is recognized in the vesting period, which is the period for fulfillment of all the defined vesting terms of the share-based payment arrangement.
At each reporting date, the Company adjusts its estimates of the number of options that are expected to vest, based on the non-market vesting conditions, and recognizes the effect of the change compared to original estimates, if any, in the statement of comprehensive loss, and a corresponding adjustment in equity.
When exercising the options, the Company issues new shares, the proceeds, net of directly attributable transaction costs, are recognized in share capital (par value) and additional paid in capital.
| P. | Segment reporting |
Operating segments are reported in a manner consistent with the internal reporting provided to the chief operating decision-maker, who is responsible for allocating resources and assessing performance of the operating segments. The Company operates in one operating segment.
| Q. | Leases |
Lease agreements in which a significant portion of the risks and rewards of ownership are retained by the lessor are classified as operating leases. Payments made in connection with operating leases are recognized in profit or loss using the straight-line basis over the term of the lease.
F-17
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 2—SIGNIFICANT ACCOUNTING POLICIES (continued)
| R. | Financial instruments: |
As of January 1, 2018, the Company adopted IFRS 9 “Financial Instruments”.
The initial adoption of IFRS 9 did not have a material effect on the consolidated financial statements of the Company.
| (a) | Financial assets | |
| 1) | Classification |
The financial assets of the Company are classified as financial assets at amortized cost. The classification is done on the basis of the Company’s business model for managing the financial asset and the contractual cash flow characteristics of the financial asset.
Financial assets at amortized cost are assets held within a business model whose objective is to hold assets in order to collect contractual cash flows and the contractual terms of the financial asset give rise on specified dates to cash flows that are solely payments of principal and interest on the principal amount outstanding.
Financial assets at amortized cost are included in current assets, except for those with maturities greater than 12 months after the statements of financial position date (for which they are classified as noncurrent assets).
Financial assets at amortized cost of the Company are included in trade receivables and other receivables in the Statements of Financial Position.
| 2) | Recognition and measurement |
Regular purchases and sales of financial assets are recognized on the settlement date, which is the date on which the asset is delivered to the Company or delivered by the Company. Investments are initially recognized at fair value plus transaction costs for all financial assets not recorded at fair value through profit or loss, except for trade receivables, that are recognized initially at the amount of consideration that is unconditional unless they contain significant financing components.
F-18
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 2—SIGNIFICANT ACCOUNTING POLICIES (continued)
Financial assets measured at fair value through profit or loss are initially recognized at fair value, related transaction costs are expensed to profit or loss. Financial assets are derecognized when the rights to receive cash flow from the investments have expired or have been transferred and the Company has transferred substantially all risks and rewards of ownership. Financial assets at fair value through profit or loss are subsequently recorded at fair value. Financial assets at amortized cost are measured in subsequent periods at amortized cost using the effective interest method.
Gains or losses arising from changes in the fair value of financial assets at fair value through profit or loss are presented in the Statement of Comprehensive Loss under “Financial Expenses (Income), net”.
As to methods for measurement of the Company’s financial instruments, see note 4.
| 3) | Impairment |
The Company recognizes a loss allowance for expected credit losses on financial assets at amortized cost.
At each reporting date, the Company assesses whether the credit risk on a financial instrument has increased significantly since initial recognition. If the financial instrument is determined to have low credit risk at the reporting date, the Company assumes that the credit risk on a financial instrument has not increased significantly since initial recognition.
The Company measures the loss allowance for expected credit losses on trade receivables that are within the scope of IFRS 15 and on financial instruments for which the credit risk has increased significantly since initial recognition based on lifetime expected credit losses. Otherwise, the Company measures the loss allowance at an amount equal to 12-month expected credit losses at the current reporting date.
Prior to the effective date and adoption of IFRS 9, the financial assets of the Company were classified into loans and receivables. The classification depended on the purpose for which the financial assets were acquired, also, prior to the adoption of IFRS 9, the Company assessed at December 31, 2017 whether there is any objective evidence that a financial asset or group of financial assets was impaired.
F-19
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 2—SIGNIFICANT ACCOUNTING POLICIES (continued)
| (b) | Financial liabilities |
| (1) | General |
Financial liabilities are initially recognized at their fair value minus, in the case of a financial liability not at fair value through profit or loss, transaction costs that are directly attributable to the issue of the financial liability.
Financial liabilities are subsequently measured at amortized cost, except for derivative financial instruments, which are subsequently measured at fair value through profit or loss (see below).
Financial liabilities are classified as current liabilities if payment is due within one year or less, otherwise they are classified as non-current liabilities.
The Company’s financial liabilities at amortized cost are included in accounts payable, accrued liabilities and other and deferred revenues.
The derivative financial instruments represent warrants that confer the right to net share settlement.
The Company removes a financial liability (or a part of a financial liability) from its statement of financial position when, and only when, it is extinguished (when the obligation specified in the contract is discharged, canceled or expired).
| (2) | Financial liabilities at fair value through profit or loss. |
Warrants, convertible debentures allotted to investors that contain an anti-dilution protection right and other rights, as well as anti-dilution derivative related to shares issued (see also Notes 12 and 14) are classified, in accordance with IAS 32 as “financial liabilities” since the number of shares that will be issued upon their settlement is not fixed. As the aforementioned liabilities are non-equity derivative financial instruments, they are measured, in accordance with IFRS 9, as financial liabilities at fair value through profit or loss.
In accordance with IFRS 9, the Company elected to designate, upon initial recognition, the entire hybrid (combined) debenture (that includes the host debenture contract and the anti-dilution protection) and warrants as a financial liability at fair value through profit or loss.
The said liabilities are measured at their fair value at each date of the balance sheet, with changes in their fair value recorded to “financial expenses (income), net” in the consolidated statements of comprehensive loss.
F-20
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 2—SIGNIFICANT ACCOUNTING POLICIES (continued)
For those liabilities for which, upon initial recognition, the transaction price is different than their fair value – the liability is initially recognized at fair value adjusted to defer the difference between the fair value at initial recognition and the transaction price (“Day 1 Loss”), as the Company uses valuation techniques that incorporate data not obtained from observable markets. After initial recognition, the unrecognized Day 1 Loss of the said liabilities is amortized on a straight line basis over the term that market participants would take into account when pricing the liability. Any unrecognized Day 1 Loss is immediately recognized in profit or loss if the fair value of the financial instrument in question can be determined either by using only market observable model inputs or by reference to a quoted price for the same product in an active market. Upon exercise of convertible debenture for which an unrecognized a Day 1 Loss exists, the carrying amount of the convertible debenture (which is presented net of the unrecognized Day 1 Loss) is reclassified to equity with no impact on profit or loss.
Transaction costs allocated to financial liabilities measured at fair value through profit or loss are recognized immediately in profit or loss.
| S. | Loss per share |
Basic loss per share is generally based on the distributable loss to ordinary shareholders, divided by the weighted average number of ordinary shares outstanding in the period, net of shares held by the Company.
When calculating diluted loss per share, the Company adjusts the loss attributable to ordinary shareholders of the Company and the weighted average number of ordinary shares outstanding, for the effects of all dilutive potential ordinary shares.
Potential shares are only taken into account if their effect is dilutive (reduces earnings per share or increases loss per share).
F-21
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 2—SIGNIFICANT ACCOUNTING POLICIES (continued)
| T. | New standards and interpretations that are not yet in effect and have not been early adopted by the Company: |
| 1) | IFRS 16 “Leases” (“IFRS 16”) |
IFRS 16 was issued in January 2016. It will result in almost all leases being recognized on the balance sheet by lessees, as the distinction between operating and finance leases is removed. Under the new standard, an asset (the right to use the leased item) and a financial liability to pay rentals are recognized. The only exceptions are short-term and low-value leases.
The Company has reviewed all of the Company’s arrangements that in effect in light of the new lease accounting rules in IFRS 16. The standard will affect primarily the accounting for the Company’s operating leases.
The Company expects to recognize lease liabilities and a right of use assets of approximately NIS 13.0 million on January 1, 2019.
The Company will apply the standard from its mandatory adoption date of January 1, 2019. The Company intends to apply the simplified transition approach and will not restate comparative amounts for the year prior to first adoption.
The Company expects that, based on the Company’s lease agreements as of December 31, 2018, net loss will increase by approximately NIS 956,000 for 2019 as a result of adopting the new rules. Operating cash flows for 2019 will increase by approximately NIS 76,000, and financing cash flows will decrease by approximately NIS 882,000 as repayment of the principal portion of the lease liabilities will be classified as cash flows from financing activities.
F-22
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 3—SIGNIFICANT ACCOUNTING ESTIMATES AND JUDGMENTS
| A. | Significant accounting estimates |
The Company makes estimates and assumptions with respect to the future. By nature, accounting estimates are rarely identical to actual results. The estimate that has a significant risk of resulting in a material adjustment to carrying amounts of assets and liabilities in the next financial year are discussed below:
| 1) | Impairment indicators of IPR&D. |
The Company reviews whether events or changes in circumstances have occurred that indicate that the carrying amount of IPR&D may not be recoverable. In such cases an impairment test is performed. See also Note 2E(1).
| 2) | Fair value measurement of debentures and warrants |
The fair value of the debentures and warrants is measured on the basis of accepted valuation models and assumptions regarding unobservable inputs used in the valuation models, see also Note 12.
| B. | Significant judgments made when applying the Company’s accounting policy: |
| 1) | Grants from the IIA |
In accordance with the accounting treatment prescribed in Note 2G, the Company’s management is required to examine whether there is reasonable assurance that the grant that was received will be repaid. In addition, if, at the date of initial recognition, the grant is recognized in the statement of comprehensive loss, the Company’s management is required to evaluate whether there is reasonable assurance of the project’s success and of payment of royalties to the IIA. The Company’s management believes that as of December 31, 2018, there is reasonable assurance that royalties will be paid to the IIA and that their present value is NIS 1.2 million. This amount was recognized as a financial liability in the statement of financial position.
| 2) | Development costs |
Development costs are capitalized in accordance with the accounting policy described in Note 2E(3). Capitalization of costs is based on management’s judgment about technological and economic feasibility. The Company’s management believes that as of December 31, 2018, the above conditions were not met, therefore development costs were not capitalized.
| 3) | Revenue recognition |
With respect to the License Agreement (see Note 13C), the Company used its judgement to identify the Company’s promises in the agreement, whether the options included provide a material right to LB and whether the promises are a distinct performance obligation. In addition, the Company uses its judgement to determine the allocation of the transaction price between its identified distinct performance obligations. The Company uses its judgement to determine that the License in the License Agreement is the predominant item to which the royalty relates. In addition, Variable Consideration consists of potential future milestone payments.
F-23
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 3—SIGNIFICANT ACCOUNTING ESTIMATES AND JUDGMENTS (continued)
The Company determined that all such Variable Consideration shall be allocated to the License (the satisfied performance obligation).
However, it will be recognized only when it would be highly probable that a significant reversal of cumulative revenues will not occur, usually upon achievement of the specific milestone.
NOTE 4—FINANCIAL INSTRUMENTS AND FINANCIAL RISK MANAGEMENT
Financial risk management:
| 1) | Financial risk factors |
The Company’s activities expose it to diverse financial risks: currency risk, credit risk, and liquidity risk. The Company’s comprehensive risk management plan focuses on the unpredictability of financial markets and the attempt to minimize potential adverse effects on the Company’s financial performance.
Risk management is carried out by the Company’s management under policies approved by the Board.
| A) | Market risks |
Exchange rate risk
The Company is exposed to exchange rate risks arising from exposure to various currencies, primarily the U.S. dollar. The exchange rate risk is due to future commercial transactions and assets or liabilities denominated in foreign currency.
As of December 31, 2018, if the Company’s Functional Currency had depreciated by 5% against the U.S. dollar, and if all the other variables had remained constant, the loss for the year would have been higher by NIS 596,000 (December 31, 2017, NIS 350,000), mainly due to losses from exchange rate differences for translation of cash balances, other receivables and trade payables.
| B) | Liquidity risk |
The Company has not yet generated profits or positive cash flows from its operating activities, and the continuation of its operations in the current format is subject to raising financing sources until a positive cash flow is generated from its operations. See also Note 1A.
| 2) | Capital risk management |
The objectives of the Company’s capital risk management are to maintain the Company’s ability to continue as a going concern in order to provide shareholders with a return on their investment and to maintain an optimal capital structure to minimize the cost of capital. See also Note 1A.
F-24
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 4—FINANCIAL INSTRUMENTS AND FINANCIAL RISK MANAGEMENT (continued)
| 3) | Estimates of fair value |
| A. | Fair value measurement |
The Company measures fair value and discloses fair value measurements for financial assets and liabilities. Fair value is based on the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. The accounting standard establishes a fair value hierarchy that prioritizes observable and unobservable inputs used to measure fair value into three broad levels, which are described below:
Level 1: Quoted prices (unadjusted) in active markets that are accessible at the measurement date for assets or liabilities. The fair value hierarchy gives the highest priority to Level 1 inputs.
Level 2: Observable inputs that are based on inputs not quoted on active markets but corroborated by market data.
Level 3: Unobservable inputs are used when little or no market data is available. The fair value hierarchy gives the lowest priority to Level 3 inputs.
In determining fair value, the Company utilizes valuation techniques that maximize the use of observable inputs and minimize the use of unobservable inputs to the extent possible and considers counterparty credit risk in its assessment of fair value.
| B. | Estimates of fair value |
The following is an analysis of the financial instruments measured at fair value, according to valuation methods. Inputs for the assets and liabilities that are not based on observable market data (unobservable inputs) (Level 3).
The Company’s financial liability at fair value through profit or loss is the obligation for warrants and anti-dilution derivatives.
F-25
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 4—FINANCIAL INSTRUMENTS AND FINANCIAL RISK MANAGEMENT (continued)
The following table presents the group’s financial liabilities measured at fair value, net of unrecognized Day 1 Loss:
| December 31, | ||||
| 2017 | ||||
| NIS in thousands |
||||
| Fair value of convertible debentures | 14,015 | |||
| Unrecognized Day 1 Loss | (1,376 | ) | ||
| Debentures, net | 12,639 | |||
| December 31, | ||||
| 2018 | ||||
| NIS in thousands |
||||
| Fair value of warrants | 4,736 | |||
| Unrecognized Day 1 Loss | (2,302 | ) | ||
| Warrants, net | 2,434 | |||
| C. | Financial instruments in level 3 |
The following table presents the Level 3 instruments roll-forward during 2017:
| 2017 | ||||
| NIS in thousands |
||||
| Opening balance as of January 1, 2017 | — | |||
| Issuance | (14,190 | ) | ||
| Profit from changes in fair value of financial instruments | 34 | |||
| Closing balance as of December 31, 2017 | (14,156 | ) | ||
The following table presents the Level 3 instruments roll-forward during 2018:
| 2018 | ||||
| NIS in thousands |
||||
| Opening balance as of January 1, 2018 | (14,156 | ) | ||
| Issuance of Warrants | (6,273 | ) | ||
| Conversion of debentures to pre-paid warrants | 13,740 | |||
| Profit from changes in fair value of financial instruments | 1,590 | |||
| Closing balance as of December 31, 2018(1) | (5,099 | ) | ||
| (1) | Represents $4,736 thousand warrants and $363 thousand anti-dilution derivatives. |
F-26
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 5—CASH AND CASH EQUIVALENTS
| December 31 | ||||||||
| 2017 | 2018 | |||||||
| NIS in thousands |
||||||||
| Breakdown by currency: | ||||||||
| NIS | 9,654 | 9,437 | ||||||
| In foreign currency (mainly U.S. dollars) | 8,163 | 10,628 | ||||||
| 17,817 | 20,065 | |||||||
NOTE 6—OTHER RECEIVABLES
| December 31 | ||||||||
| 2017 | 2018 | |||||||
| NIS in thousands |
||||||||
| Value added tax authorities | 464 | 471 | ||||||
| Receivables from IIA | 1,294 | 375 | ||||||
| Prepaid expenses | 1,717 | 393 | ||||||
| Other | 68 | 14 | ||||||
| 3,543 | 1,253 | |||||||
Most financial balances are in NIS and are unlinked.
The carrying amount of receivables is a reasonable approximation of their fair value since the effect of discounting is insignificant.
The maximum exposure to credit risk as of December 31, 2018 for receivables that are financial assets is their carrying amount. The Company does not hold any collateral for these receivables.
F-27
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 7—PROPERTY AND EQUIPMENT
Composition of property and equipment and accumulated depreciation, by principal groups, and the movements therein in 2016:
| Cost | Accumulated depreciation | |||||||||||||||||||||||||||
| Carrying amount at beginning of year |
Additions | Carrying amount at end of year |
Carrying amount at beginning of year |
Additions | Carrying amount at end of year |
Depreciated balance as at December 31, 2016 |
||||||||||||||||||||||
| NIS in thousands | NIS in thousands | NIS in thousands |
||||||||||||||||||||||||||
| Computer equipment | 662 | 40 | 702 | 569 | 54 | 623 | 79 | |||||||||||||||||||||
| Office furniture | 496 | 4 | 500 | 187 | 27 | 214 | 286 | |||||||||||||||||||||
| Laboratory equipment | 5,183 | 284 | 5,467 | 3,726 | 400 | 4,126 | 1,341 | |||||||||||||||||||||
| Greenhouse equipment | 2,982 | — | 2,982 | 2,459 | 254 | 2,713 | 269 | |||||||||||||||||||||
| Leasehold improvements | 1,043 | 1,842 | 2,885 | 813 | 39 | 852 | 2,033 | |||||||||||||||||||||
| 10,366 | 2,170 | 12,536 | 7,754 | 774 | 8,528 | 4,008 | ||||||||||||||||||||||
Composition of property and equipment and accumulated depreciation, by principal groups, and the movements therein in 2017:
| Costs | Accumulated depreciation | |||||||||||||||||||||||||||
| Carrying amount at beginning of year |
Additions | Carrying amount at end of year |
Carrying amount at beginning of year |
Additions | Carrying amount at end of year |
Depreciated balance as at December 31, 2017 |
||||||||||||||||||||||
| NIS in thousands | NIS in thousands | NIS in thousands |
||||||||||||||||||||||||||
| Computer equipment | 702 | 16 | 718 | 623 | 44 | 667 | 51 | |||||||||||||||||||||
| Office furniture | 500 | 1 | 501 | 214 | 27 | 241 | 260 | |||||||||||||||||||||
| Laboratory equipment | 5,467 | 68 | 5,535 | 4,126 | 420 | 4,546 | 989 | |||||||||||||||||||||
| Greenhouse equipment | 2,982 | — | 2,982 | 2,713 | 207 | 2,920 | 62 | |||||||||||||||||||||
| Leasehold improvements | 2,885 | 362 | 3,247 | 852 | 175 | 1,027 | 2,220 | |||||||||||||||||||||
| 12,536 | 447 | 12,983 | 8,528 | 873 | 9,401 | 3,582 | ||||||||||||||||||||||
F-28
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 7—PROPERTY AND EQUIPMENT (continued)
Composition of property and equipment and accumulated depreciation, by principal groups, and the movements therein in 2018:
| Costs | Accumulated depreciation | |||||||||||||||||||||||||||
| Carrying amount at beginning of year |
Additions | Carrying amount at end of year |
Carrying amount at beginning of year |
Additions | Carrying amount at end of year |
Depreciated balance as at December 31, 2018 |
||||||||||||||||||||||
| NIS in thousands | NIS in thousands | NIS in thousands |
||||||||||||||||||||||||||
| Computer equipment | 718 | 137 | 855 | 667 | 55 | 722 | 133 | |||||||||||||||||||||
| Office furniture | 501 | 8 | 509 | 241 | 27 | 268 | 241 | |||||||||||||||||||||
| Laboratory equipment | 5,535 | 188 | 5,723 | 4,546 | 410 | 4,956 | 767 | |||||||||||||||||||||
| Greenhouse equipment | 2,982 | - | 2,982 | 2,920 | 44 | 2,964 | 18 | |||||||||||||||||||||
| Leasehold improvements | 3,247 | 2,438 | 5,685 | 1,027 | 735 | 1,762 | 3,923 | |||||||||||||||||||||
| Vehicles | - | 212 | 212 | - | 20 | 20 | 192 | |||||||||||||||||||||
| 12,983 | 2,983 | 15,966 | 9,401 | 1,291 | 10,692 | 5,274 | ||||||||||||||||||||||
F-29
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 8—INTANGIBLE ASSETS
Composition of intangible assets and accumulated amortization, by principal groups, and the movements therein in 2016:
| Cost | Accumulated depreciation | |||||||||||||||||||||||
| Carrying amount at beginning of year |
Carrying amount at end of year |
Carrying amount at beginning of year |
Additions |
Carrying of year |
Depreciated balance as at December 31, 2016 |
|||||||||||||||||||
| NIS in thousands | NIS in thousands | NIS in thousands | ||||||||||||||||||||||
| Software | 104 | 104 | 103 | 1 | 104 | — | ||||||||||||||||||
| IPR&D | 1,720 | 1,720 | — | 89 | 89 | 1,631 | ||||||||||||||||||
| 1,824 | 1,824 | 103 | 90 | 193 | 1,631 | |||||||||||||||||||
Composition of intangible assets and accumulated amortization, by principal groups, and the movements therein in 2017:
| Costs | Accumulated amortization | |||||||||||||||||||||||
| Carrying amount at beginning of year |
Carrying amount at end of year |
Carrying amount at beginning of year |
Addition | Carrying amount at end of year |
Amortized balance as at December 31, 2017 |
|||||||||||||||||||
| NIS in thousands | NIS in thousands | NIS in thousands |
||||||||||||||||||||||
| Software | 104 | 104 | 104 | — | 104 | - | ||||||||||||||||||
| IPR&D | 1,720 | 1,720 | 89 | 177 | 266 | 1,454 | ||||||||||||||||||
| 1,824 | 1,824 | 193 | 177 | 370 | 1,454 | |||||||||||||||||||
Composition of intangible assets and accumulated amortization, by principal groups, and the movements therein in 2018:
| Costs | Accumulated amortization | |||||||||||||||||||||||
| Carrying amount at beginning of year |
Carrying amount at end of year |
Carrying amount at beginning of year |
Addition | Carrying amount at end of year |
Amortized balance as at December 31, 2018 |
|||||||||||||||||||
| NIS in thousands | NIS in thousands | NIS in thousands |
||||||||||||||||||||||
| IPR&D | 1,720 | 1,720 | 266 | 181 | 447 | 1,273 | ||||||||||||||||||
| 1,720 | 1,720 | 266 | 181 | 447 | 1,273 | |||||||||||||||||||
F-30
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 9—INCOME TAX
| A. | Taxation of the Company and its subsidiary: |
Tax rates
The income of the Company and its subsidiary is taxable at the regular rate of corporate tax in Israel.
In January 2016, the Law for the Amendment of the Income Tax Ordinance (No. 216) was published, enacting a reduction of corporate tax rate beginning in 2016, from 26.5% to 25%.
In December 2016, the Economic Efficiency Law (Legislative Amendments for Achieving Budget Objectives in 2017 and 2018), 2016 was published. The Law stipulates a further reduction in the rate of corporate tax, from 25% to 23%. However, the Law establishes a temporary order by which the corporate tax rate in 2017 will be 24%. As a result, the corporate tax rate applicable in 2017 was 24% and the corporate tax rate from 2018 onwards is 23%. The changes in the above tax rates have no effect on the Company’s financial statements.
The corporate tax rate for 2018, 2017 and 2016 was 23%, 24% and 25%, respectively.
| B. | Carry-forward tax losses |
Deferred tax assets for carry-forward tax losses are recognized if it is probable that the tax benefit will be realized through the existence of future taxable profits.
The carry-forward losses of CollPlant Holdings Ltd. (without capital losses) as at December 31, 2018 and 2017 amounted to approximately NIS 12.6 million and NIS 11.1 million, respectively.
The carry-forward losses of CollPlant Ltd. (without capital losses) as at December 31, 2018 and 2017 amounted to approximately NIS 152 million and NIS 148 million, respectively.
Under Israeli tax laws, carryforward tax losses have no expiration date.
The Company did not recognize deferred taxes on the losses of the Company and the subsidiary, as it is not probable that the carry-forward losses will be realized in the foreseeable future.
| C. | Tax assessments |
In accordance with the Israeli Income Tax Ordinance, tax assessments filed by the Company and its subsidiary up to 2013 are considered final.
| D. | Value added tax |
The Company and its subsidiary are registered as authorized dealers in Israel for VAT purposes.
F-31
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 10—ACCOUNTS PAYABLE
| December 31 | ||||||||
| 2017 | 2018 | |||||||
| NIS in thousands | ||||||||
| A. Trade payables: | ||||||||
| Breakdown by currency: | ||||||||
| NIS | 1,923 | 1,656 | ||||||
| In foreign currency (mainly U.S. dollars) | 999 | 675 | ||||||
| 2,922 | 2,331 | |||||||
| B. Composition of other payables: | ||||||||
| Employees and institutions for employees | 1,148 | 1,445 | ||||||
| Provisions for vacation and others | 658 | 734 | ||||||
| Other | 190 | 187 | ||||||
| 1,996 | 2,366 | |||||||
The carrying amount of accounts payable is a reasonable approximation of their fair value since the effect of discounting is insignificant.
NOTE 11—RETIREMENT BENEFIT OBLIGATION
The amount recognized as an expense for defined contribution plans in 2016, 2017 and 2018 is NIS 1,558 thousand NIS 1,378 thousand and NIS 1,627 thousand, respectively.
NOTE 12—FINANCING AGREEMENT
On September 6, 2017, the Company signed a securities purchase agreement (the “Alpha Purchase Agreement”) with Alpha Capital Anstalt (“Alpha”) , pursuant to which the Company agreed, upon the terms and subject to the conditions of the Alpha Purchase Agreement, to issue to Alpha, in a private placement, certain securities, in three tranches, as follows: (i) at the first closing, which was completed on October 26, 2017, ordinary shares and a Convertible Debenture (“Debenture”), for a purchase price of $2 million, (ii) at the second closing, which was completed on December 31, 2017 and which was subject, among other things, to approval of the private placement by the Company’s shareholders, a Debenture for a purchase price of $2 million, and (iii) at the third closing, which was completed on April 30, 2018, which was subject, among other things, to the listing of the Company’s ADSs for trading on the NASDAQ and to the receipt of shareholder and option holder approval to adopt the provisions of Chapter E3 of the Israeli Securities Law of 1968 (which allows the Company to report in Israel in accordance with U.S. reporting requirements) (“Dual Reporting Approval”), ordinary shares and/or a Debenture for a purchase price of $1 million, and a warrant (the “Alpha Warrant”) to purchase 49,607,407 ordinary shares represented by 992,148 ADSs exercisable for a period of five years from the date of issuance at an exercise price of the US dollar equivalent of NIS 36.14379 per ADS (calculated in accordance with the known representative rate of exchange on the date of the notice of exercise).
F-32
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 12—FINANCING AGREEMENT (continued)
On October 26, 2017, upon the completion of the first closing, the Company issued to Alpha 7,280,000 ordinary shares and a Debenture in the principal amount of $1,375,144, for gross proceeds of $2,000,000. On December 31, 2017, upon completion of the second closing, the Company issued a Debenture in the principal amount of $2,000,000 for gross proceeds of $2,000,000. The Debentures were convertible at any time at the option of the holder into ADSs at a conversion price of the US dollar equivalent of NIS 15.3897 (calculated in accordance with the rate of exchange of NIS 3.586 per $1.00) per ADS. In addition, the Debenture was mandatorily convertible at the then effective conversion price without regard to any beneficial ownership limitation if (i) the ADSs or the Company’s ordinary shares are approved for listing on the Nasdaq stock market, and (ii) certain equity conditions are met, and provided that the holder may elect to convert the Debenture in whole or in part to a pre-paid warrant to purchase such number of ADSs otherwise issuable upon mandatory conversion of the Debenture. On January 31, 2018, Debentures in the aggregate principal amount of $3,375,144 were automatically converted into a pre-paid warrant to purchase 39,322,742 ordinary shares represented by 786,455 ADSs.
On April 30, 2018, the Company completed the third closing of the Alpha Purchase Agreement, which resulted in the issuance to Alpha of a pre-paid warrant to purchase 9,921,482 ordinary shares represented by 198,430 ADSs and the Alpha Warrant to purchase up to 49,607,407 ordinary shares represented by 992,149 ADSs, at an exercise price of NIS 36.14 per ADS ($10.28 per ADS), for gross proceeds of $1 million. As the Alpha Warrant includes an anti-dilution protection and other rights, they are classified as financial liabilities measured at fair value through profit or loss at each reporting period.
In 2018, Alpha converted a pre-paid warrant to purchase 8,250,000 ordinary shares into 8,250,000 ordinary shares represented by 165,000 ADSs.
As part of the first and second closings, and included within the ordinary shares and Debentures issued at the first and second closings, the Company issued an aggregate of 1,080,503 ordinary shares and Debentures convertible into 5,836,313 ordinary shares in connection with services Alpha provided to the Company. These issuances, having a fair market value of NIS 3.0 million, were accounted as share based compensation. NIS 1.5 million was recognized as an expense within “general, administrative and marketing expenses” in the statements of comprehensive loss in 2017 and 2018, respectively.
Under the Alpha Purchase Agreement, Alpha was also granted certain rights, including, among other things, anti-dilution protection until October 26, 2019 in the event of certain subsequent equity issuances at a price that is lower than the then applicable per ordinary share purchase price. This right is accounted for as a derivative liability. Accordingly, it is presented as a component of non-current liabilities and is measured at fair value each reporting period and presented in the statements of financial position within non-current liabilities, see Note 2R.
F-33
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 12—FINANCING AGREEMENT (continued)
The Warrants are initially recognized at fair value adjusted to defer the difference between the fair value at initial recognition and the transaction price (“Day 1 Loss”), as the Company uses valuation techniques that incorporate data not obtained from observable markets.
As for the accounting treatment of the Day 1 Loss - see Note 2R.
The financial instruments recognized on the Company’s statements of financial position as of December 31, 2017 and 2018, are as follows:
1) Derivatives – comprised of an anti-dilution protection on 37,320,978 and 27,399,497 ordinary shares as of December 31, 2018 and 2017, respectively. The derivative is presented in the Company’s balance sheets on a fair value basis in the amount of NIS 363,186 and 140,875 as of December 31, 2018 and 2017, respectively.
The derivative fair value is determined by using a put option Model.
The following table presents the assumptions that were used for the models as of December 31, 2017:
| Alpha | Meitav Dash and Ami Sagi* | |||||||
| Probability | 5 | % | 5 | % | ||||
| Expected volatility | 64.35 | % | 65.64 | % | ||||
| Risk free interest rate | 0.185 | % | 0.187 | % | ||||
| Expected term (years) | 2 | 2 | ||||||
The following table presents the assumptions that were used for the models as of December 31, 2018:
| Alpha | Meitav Dash and Ami Sagi* | |||||||
| Probability | 5 | % | 5 | % | ||||
| Expected volatility | 57.33 | % | 53.31 | % | ||||
| Risk free interest rate | 0.47 | % | 0.55 | % | ||||
| Expected term (years) | 0.82 | 0.99 | ||||||
| * | See Note 14(A)(10) and (11) |
F-34
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 12—FINANCING AGREEMENT (continued)
2) Warrants - to purchase up to 49,607,407 ordinary shares and include an anti-dilution protection right and other rights as of December 31, 2018. The warrants are presented in the Company’s statement of financial position at fair value basis in the amount of NIS 2,434 thousand, net of unrecognized Day 1 Loss in the amount of NIS 2,302 thousands, see note 4.
The warrants fair value is determined by using the B&S model.
The following table presents the assumptions that were used for the model as of December 31, 2018:
| Warrants | ||||
| Fair value of Warrant– NIS | 0.0955 | |||
| Expected volatility | 63.79 | % | ||
| Risk free interest rate | 1.47 | % | ||
| Expected term (years) | 4.33 | |||
| Expected dividend yield | 0 | % | ||
3) Debentures - convertible debentures which are convertible into 39,322,742 prepaid warrants and contain an anti-dilution protection right and other rights as of December 31, 2017. The Debentures are presented in the Company’s statement of financial position at fair value basis in the amount of NIS 12,639 thousand, net of unrecognized Day 1 Loss in the amount of NIS 1,376 thousand.
The Debentures’ fair value is determined by using an Asian put option model.
The following table presents the assumptions that were used for the models as of December 31, 2017:
| First closing |
Second closing | |||||||
| Debenture | Debenture | |||||||
| Fair value of shares of common stock - NIS | 0.43 | 0.43 | ||||||
| Expected volatility | 64.56 | % | 61.29 | % | ||||
| Discount on lack of marketability | 20.4 | % | 20.2 | % | ||||
| Risk free interest rate | 0.17 | % | 0.19 | % | ||||
| Expected term (years) | 1.82 | 2 | ||||||
| Expected dividend yield | 0 | % | 0 | % | ||||
F-35
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 13—COMMITMENTS AND CONTINGENT LIABILITIES
| A. | Agreements: |
| 1) | Operating lease agreements: |
| A) |
On November 15, 2018, the Company signed a new lease agreement for the Company’s new offices located in Rehovot. The lease is for five years and five months in return for a monthly payment of NIS 89 thousand, with an option to extend for five additional years. In addition, as part of the lease agreement the Company will not carry the rent monthly payment during the first five months of the lease agreement and will be reimbursed for its building adjustments costs in the amount of NIS 2,500 thousand.
As collateral for the lease agreement, a restricted deposit was pledged in favor of the property owner. The balance of the restricted deposit as of December 31, 2018 amounts to NIS 580 thousand. The deposit is classified as a non-current asset.
|
|
| B) | In 2017, an agreement was signed to extend the lease of the Company’s offices, which commenced in June 2008. The lease term ended on August 18, 2018, and the Company signed an amendment to extend the lease until March 31, 2019. The monthly rent amounts to NIS 54 thousand. |
As collateral for the lease agreement, a restricted deposit was pledged in favor of the property owner. The balance of the restricted deposit as of December 31, 2018 amounts to NIS 544 thousand. The deposit is classified as current asset.
| C) | In April 2007, the Company signed an agreement with a third party for lease of land in Yessod Hamaala. The lease term ended on April 30, 2017. On July 4, 2017, the Company signed a new agreement for four years with an option for extension of another 6 years. The lease term began on May 1, 2017. The annual rent amount is NIS 120 thousand. |
| D) | On July 28, 2016, the Company signed a lease agreement for additional space designated for its development and production activities. The lease is for three years with an option to extend for four additional years, in return for a monthly payment of NIS 30 thousand. In addition, as part of the lease agreement, the Company acquired equipment and clean rooms for the Company’s operations for NIS 1,849 thousand. Out of the aforementioned total consideration an amount of NIS 1,197 thousand was paid by issuing 1,067,916 ordinary shares of the Company and a total of NIS 525 thousand was on credit and will be repaid in cash over the term of the lease. The Company intends to exercise its first option to extend the lease period. |
F-36
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 13—COMMITMENTS AND CONTINGENT LIABILITIES (continued)
| 2) | Commitment to pay royalties to the Government of Israel |
The Company is committed to pay royalties to the Government of Israel on proceeds from sales of products in the research and development of which the Government participates by way of grants through the IIA.
At the time the grants were received, successful development of the related project was not assumed. In the case of failure of the project that was partly financed by the Government of Israel, the Company is not obligated to pay any such royalties. Under the terms of Company’s funding from the Israeli Government, royalties of 3%-3.5% are payable on sales of products developed from projects so funded up to 100% of the amount of the grant received by the Company (dollar linked) with the addition of an annual interest based on Libor.
The updated estimate of the Company as of December 31, 2018 is that royalties will be paid to the IIA and that their present value is NIS 1,228 thousand. This amount was recognized as a financial liability in the statement of financial position (NIS 1,185 thousand within long-term liabilities, and the remainder within current liabilities). As of December 31, 2018, the fair value of that liability is not materially different from its carrying amount. As of December 31, 2018, the maximum royalty amount that would be payable by the Company, before additional Libor interest, is approximately NIS 31.8 million (assuming 100% of the funds are payable).
During 2018, grants amounting to NIS 2.2 million were received from the IIA. The participation of IIA in research and development expenses is presented net of expenses to pay royalties and remeasurement of the IIA liability and amounted to NIS 3.2 million.
| B. | Development agreements with pharmaceutical and orthobiologic companies |
On November 17, 2010, CollPlant Ltd. and Pfizer signed an agreement for joint development of prototype products for the treatment of orthopedic problems. The agreement provided for, among other things, the allocation of the rights of the project outcomes. In accordance with the agreement, Pfizer paid CollPlant immaterial amounts for the development of prototypes.
On December 22, 2011, CollPlant and Pfizer signed another joint development agreement for development of a product for the orthopedic market (the “Development Agreement”). In accordance with the Development Agreement, the parties agreed to collaborate in the development of a product that contained Pfizer’s therapeutic proteins and compounds based on CollPlant’s recombinant human collagen (rhCollagen) (the “Product”).
F-37
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 13—COMMITMENTS AND CONTINGENT LIABILITIES (continued)
To the best of the Company’s knowledge, based partially on public sources, in July 2013, Pfizer signed an agreement with Bioventus LLC, a U.S. based company (“Bioventus”), which specializes in orthobiologics, whereby Pfizer granted Bioventus an exclusive, global license for the portfolio of projects related to Pfizer’s bone morphogenetic protein (“BMP”). Between July 2013 and February 2017, the Company and Bioventus developed a bioactive implant for spinal fusion and orthopedic trauma, instead of under the Pfizer agreement, which expired during 2014.
On July 9, 2015, the Company signed a non-binding term sheet with Bioventus. According to the term sheet, Bioventus agreed to make payments to the Company for the full development plan.
On March 1, 2017, Bioventus informed the Company that it had decided to discontinue the joint development with CollPlant and to complete product development at a subsidiary of Bioventus.
| C. | LICENSE DEVELOPMENT AND COMMERCIALIZATION AGREEMENTS |
On October 19, 2018, CollPlant entered into the License Agreement with LB, a public benefit corporation and wholly-owned subsidiary of United Therapeutics Corporation, pursuant to which LB will be entitled to develop engineered lungs or lung substitutes using CollPlant’s rhCollagen and BioInk.
Pursuant to the License Agreement, CollPlant granted to LB and its affiliates, an exclusive, perpetual, royalty-bearing and transferable license of CollPlant’s technology relating to the production and use of rhCollagen and BioInk for the commercialization of engineered lungs or lung substitutes using 3D bioprinting processes throughout the universe.
Further, under the License Agreement, CollPlant granted to LB and its affiliates, a two-year option to extend the license to engineered organs or organ substitutes of up to three additional organs specified in the License Agreement (each an “Option Product” and together with lungs, the “Covered Products”). Further, at the end of the option period, LB and its affiliates shall have a one-year right of first refusal to receive an exclusive license under CollPlant’s technology relating to the production and use of rhCollagen and BioInk for the Option Products. Other than under the license Agreement, CollPlant has agreed not to conduct, enable or fund any research, development or commercialization, or grant any license, with respect to the Covered Products during the term of the License Agreement, unless with respect to any Option Product, the option is not exercised and the right of first refusal period expires.
The License Agreement provides that LB will purchase CollPlant’s BioInk on a non-exclusive basis for use in the development and manufacture of Covered Products and for up to the first two years of the License Agreement, CollPlant will supply LB with a specified limited quantity of BioInk without charge. The License Agreement further provides that following effectiveness, LB will build a facility, or engage a manufacturer to build a facility, in the U.S. for the manufacture of the Company’s rhCollagen and BioInk and the parties have agreed that LB has the option to purchase consulting services for the design of the facility.
F-38
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 13—COMMITMENTS AND CONTINGENT LIABILITIES (continued)
The License Agreement provides for the payment of an upfront cash payment of $5 million to CollPlant, which was paid to CollPlant in November 2018 following effectiveness of the Agreement. In addition, the License Agreement provides for a one-time non-refundable option payments of $3 million per Option Product ($9 million in the aggregate), and up to $30 million of milestone payments payable as follows: (i) $5 million upon completion of the U.S. facility design, (ii) $5 million upon completion of production of a specified amount of BioInk, and (iii) $5 million for FDA marketing approval for each Covered Product (up to $20 million in the aggregate). Further, CollPlant shall be entitled to a fixed-fee royalty payment (subject to certain adjustment) for each product commercially sold during the term of the License Agreement, a fee for the supply of certain quantities of BioInk to LB, and reimbursement for certain costs related to the U.S. facility and any payment made by CollPlant to the IIA.
Unless earlier terminated, the License Agreement will continue in effect on a Covered Product-by-Covered Product and country-by-country basis until the later of (i) the expiration, invalidation or abandonment of the last CollPlant patent covering a Covered Product in a particular country, and (ii) 12 years from the first commercial sale of such Covered Product in such country. Following expiration (unless earlier terminated), the licenses granted to LB in the License Agreement will continue on a fully paid-up, irrevocable basis. The License Agreement may be terminated early by either party for material breach or bankruptcy. In addition, CollPlant may terminate the License Agreement in the case of a challenge made by LB, its affiliates or sub-licensees with respect to a CollPlant patent covering a Covered Product or if LB and its affiliates and sub-licensees discontinue development and commercialization of all Covered Products for at least one year. LB may terminate the License Agreement at any time upon 30 days’ written notice with respect to the entirety of the License Agreement and upon 30 days’ written notice with respect to its license and other rights under the License Agreement relating to one or more CollPlant patents, on a patent by- patent and country-by-country basis.
| D. | Contingent liability |
On September 6, 2017, the Company received a VAT assessment from the Israel Tax Authority according to which the Company is required to pay tax in the amount of NIS 1.5 million (including linkage differentials and interest) for the years 2012-2016. The Company disputed the position of the Israel Tax Authority resulting in the latter to increase its VAT assessment and require the Company to pay tax in the amount of NIS 1.8 million (including linkage differentials and interest) for the abovementioned period. The Company has appealed the entire assessment to the District Court, in view of its position that it is not liable for the entire tax requirement. A preliminary hearing in the District Court in Lod is scheduled for May 2019. The Company’s position relies, among other things, on an agreement signed between the Company and the Israel Tax Authority in 2011, which allows the Company to deduct VAT as stated. It is management’s view that its financial statements include an adequate provision in respect of the above.
F-39
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 14—EQUITY
| A. | Ordinary shares and warrants: |
| 1) | Composition |
| Number of shares | ||||||||||||||||
| Registered | Issued and paid up | |||||||||||||||
| December 31 | December 31 | |||||||||||||||
| 2018 | 2017 | 2018 | 2017 | |||||||||||||
| Ordinary shares of par value NIS 0.03 | 750,000,000 | 500,000,000 | 190,735,668 | 166,816,328 | ||||||||||||
| Amount in NIS | ||||||||||||||||
| Registered | Issued and paid up | |||||||||||||||
| December 31 | December 31 | |||||||||||||||
| 2018 | 2017 | 2018 | 2017 | |||||||||||||
| Ordinary shares of par value NIS 0.03 | 22,500,000 | 15,000,000 | 5,722,070 | 5,004,490 | ||||||||||||
Traded on NASDAQ as of January 31, 2018.
On January 31, 2018 the Company’s ADSs commenced trading on the Nasdaq Capital Market, under the symbol CLGN. Each ADS represents 50 ordinary shares.
On October 29, 2018, the Company’s ordinary shares delisted from the Tel Aviv Stock Exchange (“TASE”). See note 14A(15).
The above table does not include 920,461 shares held by the Company. These shares are considered to be dormant.
| 2) | The ordinary shares confer on their holders the right to vote and participate in shareholder meetings (with one vote for each share), the right to receive profits and the right to participate in surplus assets on liquidation of the Company. |
| 3) |
In 2016, Series F and J warrants expired without exercise.
In 2018, Series G and H warrants expired without exercise. |
| 4) | On February 2, 2016, the Company completed a capital raise of NIS 8.2 million in gross proceeds to two institutional investors and to the public (the issuance expenses amounted to NIS 643 thousand). In consideration, the Company issued 5,745,903 ordinary shares, 12,930,505 Series I warrants exercisable into 4,310,168 ordinary shares at an exercise price of NIS 0.80 per warrant, for three years, and 8,618,855 Series J warrants exercisable into 2,872,952 ordinary shares at an exercise price of NIS 0.575 per warrant, exercisable until July 31, 2016. In addition, under the terms of the broker agreement, the Company issued to the Israeli broker 814,520 Series I warrants exercisable into 271,507 ordinary shares at an exercise price of NIS 0.80 per warrant, for three years. On July 31, 2016, 8,618,855 Series J warrants expired. |
F-40
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 14—EQUITY (continued)
| 5) | On June 9, 2016, the Company completed a capital raise of NIS 11.8 million in gross proceeds by way of a non-uniform offering to institutional investors and a uniform offer to the public (the issuance expenses amounted to NIS 684 thousand). In consideration, the Company issued 11,267,833 ordinary shares and 33,803,500 Series K warrants exercisable into 11,267,833 ordinary shares at an exercise price of NIS 0.60 per warrant, for three years. In addition, in consideration, the Company issued to the broker under the terms of the broker agreement, 2,728,000 Series K warrants exercisable into 909,333 ordinary shares at an exercise price of NIS 0.60 per warrant, for three years. |
| 6) | On July 28, 2016, as part of the lease agreement described in note 13A(1)(D), the Company acquired equipment and clean rooms for the Company’s operations for NIS 1,849 thousand (present value). Of this amount, NIS 1,197 thousand was paid by issuing 1,067,916 ordinary shares and a total of NIS 525 thousand was a credit that will be repaid in cash over the term of the lease. |
| 7) | On November 17, 2016, the general meeting of shareholders approved a reverse share split of the Company’s shares that was effected on November 20, 2016. Pursuant to the reverse split each 3 ordinary shares of NIS 0.01 par value were converted into one share of NIS 0.03 par value of the Company. |
Additionally, according to the share option plan of the Company, every 3 unlisted options that were allocated through private offers to directors, employees, consultants and officers under the option plan are exercisable into one ordinary share of the Company of NIS 0.03 par value. No change took place in the exercise price of the options, as above; however, the total exercise price for one share of NIS 0.03 par value will be the former exercise price for one share of NIS 0.01 par value multiplied by 3.
Further, according to the terms and conditions of the marketable warrants of the Company, each 3 marketable warrants that the Company issued are exercisable into one ordinary share of the Company of NIS 0.03 par value. There will be no change in the exercise price of those warrants; however, the total exercise price for one share of NIS 0.03 par value will be the former exercise price for one share of NIS 0.01 par value multiplied by 3.
Following the reverse split, the Company retrospectively reflected the change in the share capital of the Company for all periods presented. Unless otherwise indicated, all of the share numbers, losses per share, share prices, options and warrants in these financial statements have been adjusted, on a retroactive basis, to reflect this 1 to 3 reverse share split.
F-41
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 14—EQUITY (continued)
| 8) | On February 12, 2017, the Company completed a capital raise of NIS 7.2 million in gross proceeds from institutional investors and from the public (the issuance expenses amounted to NIS 404 thousand). In consideration, the Company issued 21,152,000 ordinary shares and 10,576,000 Series L warrants exercisable into 10,576,000 ordinary shares at an exercise price of NIS 0.36 per warrant, until June 13, 2017. In addition, under the terms of the broker agreement, the Company issued to the broker 941,400 Series L warrants exercisable into 941,400 ordinary shares at an exercise price of NIS 0.36 per warrant. |
| 9) | During the second quarter of 2017, 10,055,464 Series L warrants were exercised into 10,055,464 ordinary shares, at an exercise price of NIS 0.36 for each warrant. The total consideration amounted to NIS 3,618 thousand. 1,461,936 Series L warrants that were not exercised expired on June 14, 2017. |
| 10) | On November 8, 2017, the Company signed a securities purchase agreement (the “Meitav Dash Purchase Agreement”) with Meitav Dash, a company held by Meitav Dash Ltd., one of the Company’s shareholders pursuant to which the Company agreed, upon the terms and subject to the conditions of the Meitav Dash Purchase Agreement, to issue to Meitav Dash in a private placement certain securities in three tranches as follows: (i) at the first closing, which was completed on December 26, 2017, 9,500,000 ordinary shares, for a purchase price of NIS 3.8 million, (ii) at the second closing, which was completed on December 26, 2017, 2,400,000 ordinary shares for a purchase price of NIS 960 thousand provided that Meitav Dash shall not be obligated to buy or hold, immediately following the second closing, 20% or more of the Company’s share capital, and (iii) at the third closing, which was completed on March 7, 2018 and which was subject, among other things, to the listing of the Company’s ADSs for trading on the Nasdaq and Dual Reporting Approval, for no additional consideration, warrants exercisable into 11,900,000 ordinary shares. |
The Company completed the first and second closings on December 26, 2017 which resulted in the issuance to Meitav Dash of an aggregate of 11,900,000 ordinary shares for gross proceeds of NIS 4,760,000 ($1,384,824) and on March 7, 2018, the Company completed the third closing which resulted in the issuance to Meitav Dash of a warrant to purchase 11,900,000 ordinary shares.
The warrant may be exercised for a period of five years from issuance at an exercise price of the US dollar equivalent of NIS 40 per ADS (calculated in accordance with the known representative rate of exchange on the date of the notice of exercise).
Under the Meitav Dash Purchase Agreement, Meitav Dash was also granted certain rights, including, among others, anti-dilution protection in the event of certain subsequent equity issuances at a price that is lower than the then applicable per ordinary share purchase price. This component is accounted for derivatives and presented in the statements of financial position within non-current liabilities, see Note 2R and Note 12.
F-42
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 14—EQUITY (continued)
| 11) | On November 9, 2017, the Company signed a securities purchase agreement (the “Sagy Purchase Agreement”) with Ami Sagy, one of the Company’s shareholders, pursuant to which the Company agreed, upon the terms and subject to the conditions of the Sagy Purchase Agreement, to issue to Ami Sagy in a private placement certain securities in two tranches as follows: (i) at the first closing, which closed on December 26, 2017, 9,300,000 ordinary shares, for a purchase price of NIS 3.7 million, and (ii) at the second closing, which closed on March 7, 2018 and which was subject, among other things, to the listing of the Company’s ADSs for trading on the Nasdaq and to Dual Reporting Approval, for no additional consideration, the Company will issue warrants exercisable into 9,300,000 of its ordinary shares. |
The Company completed the first closing on December 26, 2017 which resulted in the issuance to Ami Sagy of an aggregate of 9,300,000 ordinary shares for gross proceeds of NIS 3,720,000 ($1,054,122) and on March 7, 2018, the Company completed the second closing which resulted in the issuance to Ami Sagy of a warrant to purchase 9,300,000 ordinary shares.
The warrant may be exercised for a period of five years from issuance at an exercise price of the US dollar equivalent of NIS 40 per ADS (calculated in accordance with the known representative rate of exchange on the date of the notice of exercise).
Under the Sagy Purchase Agreement, Ami Sagy was also granted certain rights, including, among other things, anti-dilution protection in the event of certain subsequent equity issuances at a price that is lower than the then applicable per ordinary share purchase price. This component is accounted for derivatives and presented in the statements of financial position within non-current liabilities, see Note 2R.
| 12) | On September 6, 2017, the Company signed a securities purchase agreement with Alpha, see note 12. |
| 13) | On January 18, 2018, the Company signed Security Purchase Agreements for the purchase and sale, in a private placement, of an aggregate of 4,344,340 ordinary shares for an aggregate of NIS 2.2 million to the following three investors as follows: (i) Alpha entered into a Security Purchase Agreement for the purchase of 1,275,340 ordinary shares for NIS 638 thousand; (ii) Ami Sagi entered into a Security Purchase Agreement for the purchase of 2,046,000 ordinary shares for NIS 1 million; and (iii) Docor International BV entered into a Security Purchase Agreement for the purchase of 1,023,000 ordinary shares for NIS 511 thousand. Closing occurred on January 25, 2018. |
| 14) | On March 1, 2018, an extraordinary general meeting of the shareholders of the Company, approved the increase of the authorized share capital of the Company by 250,000,000 ordinary shares to 750,000,000 ordinary shares, par value NIS 0.03 per share. |
F-43
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 14—EQUITY (continued)
| 15) |
On March 20, 2018 the board of directors resolved to delist all of Company’s securities from trading on the TASE. In accordance with the Israeli Securities Law and the rules of the TASE, as the Company had four different series of warrants traded on the TASE, in order to effectuate the resolution, the Company was required to enter into an arrangement between the Company, shareholders and the holders of warrants, pursuant to Section 350 of the Israeli Companies Law.
On April 16, 2018, the Company petitioned the District Court of Lod, Israel, or the Court, to approve the convening of a shareholders’ meeting and meetings of holders of Series I Warrants and Series K Warrants, in order to approve an arrangement for the delisting of all of Company securities from TASE and the reduction of the exercise price of Series I and Series K Warrants to NIS 0.4 each (the “Arrangement”). The holders of Series G Warrants and Series H Warrants were not part to the Arrangement as such warrants expire before the expected date of the delisting of the Company’s securities from the TASE. On July 29 2018, the Court approved the Arrangement, following its approval by the different meetings of shareholders and holders of Series I and Series K Warrants. The last date of trading of the ordinary shares, Series I Warrants and Series K Warrants on the TASE was on October 29, 2018. |
| 16) | On July 11, 2018, following the Company’s board of directors and shareholders’ approval, the Company issued to Alpha a pre-paid warrant to purchase 1,060,000 ordinary shares represented by 21,200 ADSs, in connection with services Alpha provided to the Company. The issuance in fair market value of NIS 494 thousands was accounted as share based compensation and recognized as an expense within “general, administrative and marketing expenses” in the statements of comprehensive loss. |
| 17) | On July 26, 2018, the Company entered into a Securities Purchase Agreement with an investor, pursuant to which the Company issued on July 31, 2018, in a private placement, 11,125,000 ordinary shares for an aggregate purchase price of NIS 4,561,250 (approximately $1.25 million). |
F-44
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 14—EQUITY (continued)
| B. | Share-based payment: |
In accordance with an option plan for employees and consultants (the “Option Plan”), as amended from time to time, employees and consultants of the Company will be granted options, each exercisable into one ordinary share of the Company of NIS 0.03. The ordinary shares that will be issued in accordance with the Option Plan will have the same rights as the other ordinary shares of the Company, immediately subsequent to their issue. An option that is not exercised within 10 years from the allotment date will expire, unless the board of directors extends its validity.
Grants to employees are made in accordance with the Option Plan, and are carried out within the provisions of Section 102 of the Israel Income Tax Ordinance. In accordance with the track selected by the Company and these provisions, the Company is not entitled to claim a tax deduction for the employee benefits.
For those who are not employees of the Company, and for the Company’s controlling shareholders (as defined in the Income Tax Ordinance) options are granted in accordance with section 3(I) of the Income Tax Ordinance.
| 1) | On August 22 2017, the general meeting of shareholders approved the grant of 486,000 options to one director, exercisable into 486,000 shares in two tranches. 221,000 options were granted without an exercise price and vested immediately on the grant date. The fair value of each of these latter options is NIS 0.29 and is equal to the share price at the date of grant. The remaining 265,000 options are exercisable at an exercise price of NIS 0.33 per option. The options will vest over four years in which one quarter will vest one year after the grant date and the remaining balance will vest in equal parts at the end of each subsequent quarter. The fair value of each option, at the grant date, calculated according to the Black and Scholes formula, amounted to NIS 0.13. This value is based on the following assumptions: expected dividend at a rate of 0%, expected volatility at a rate of 60.53%, risk-free interest rate of 2%, and 4 years expected term. The fair value of the grant as calculated on the date of the shareholders’ approval is NIS 99 thousand. |
| 2) | On December, 2017, the board of directors approved the grant of an aggregate of 9,100,000 options to purchase 9,100,000 ordinary shares to certain officers and employees. Each of the foregoing options may be exercised at a price per option of NIS 0.58 and the options will vest over four years in which one quarter will vest one year after the grant date and the remaining balance will vest in equal parts at the end of each subsequent quarter. |
The options will vest over four years in which one quarter will vest one year after the grant date and the remaining balance will vest in equal parts at the end of each subsequent quarter.
The fair value of each option, at the grant date, calculated according to the Black and Scholes formula, amounted to NIS 0.23. This value is based on the following assumptions: expected dividend at a rate of 0%, expected volatility at a rate of 61.96%, risk-free interest rate of 2%, and 4 years expected term. The fair value of the grant as calculated at the grant date was NIS 2,145 thousand.
F-45
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 14—EQUITY (continued)
| 3) |
On January 14, 2018, the Company’s shareholders approved (i) the grant of 3,750,000 options to purchase 3,750,000 ordinary shares to Yehiel Tal, the chief executive officer, (ii) the grant of 650,000 options to purchase 650,000 ordinary shares to Adi Goldin, a director and former chairman, (iii) the grant to each of the directors, Abraham Havron, David Tsur and Scott Burell, of 500,000 options to purchase 500,000 ordinary shares, (iv) the grant to each of Gili Hart, external director, and Elan Penn, external director, of 500,000 options to purchase 500,000 ordinary shares, and (v) the annual and attendance compensation to David Tsur, in accordance with the fixed amounts in accordance with the Companies Law. Following their approval, each of the foregoing options may be exercised at a price per option of NIS 0.58 and the options will vest over four years, in which one quarter will vest one year after the grant date and the remaining balance will vest in equal parts at the end of each subsequent quarter.
The fair value of each option, at the grant date, calculated according to the Black and Scholes formula, amounted to NIS 0.29. This value is based on the following assumptions: expected dividend at a rate of 0%, expected volatility at a rate of 63%, risk-free interest rate of 2%, and 4 years expected term. The fair value of the grant as calculated at the grant date was NIS 1,998 thousand. |
| 4) | On March 20, 2018, the board of directors approved the grant of an aggregate of 900,000 options to purchase 900,000 ordinary shares to one of the company’s officers. Each of the foregoing options may be exercised at a price per option of NIS 0.49 and the options will vest over four years in which one quarter will vest one year after the grant date and the remaining balance will vest in equal parts at the end of each subsequent quarter. |
The fair value of each option, at the grant date, calculated according to the Black and Scholes formula, amounted to NIS 0.19. This value is based on the following assumptions: expected dividend at a rate of 0%, expected volatility at a rate of 62.87%, risk-free interest rate of 2%, and 4 years expected term. The fair value of the grant as calculated at the grant date was NIS 170 thousand.
| 5) | On May 1, 2018, the board of directors approved the grant of an aggregate of 250,000 options to purchase 250,000 ordinary at an exercise price of NIS 0.44 per option to a member of the scientific advisory board. |
| 6) |
On September 10, 2018, the Company signed a one year service agreement with a service provider according to which in return to its services the Company will pay a monthly retainer and issue a total of 12,000 restricted ADSs (600,000 ordinary shares) in 3 tranches of 4,000 ADSs (200,000 ordinary shares) each: (i) following the execution of the agreement, (ii) Febuary 1, 2019, (iii) June 1, 2019. If the agreement is canceled prior to the issuance date the share balance is not owed.
The first tranche was completed on December 19, 2018. |
F-46
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 14—EQUITY (continued)
Exercise of options
| 7) | On August 15, 2016, 3,620,885 options were exercised for 235,413 ordinary shares of the Company. No cash was received for the exercise. |
Changes in number of options and weighted average exercise prices in NIS are as follows:
| Year ended December 31, 2016 |
Year ended December 31, 2017 |
Year ended December 31, 2018 |
||||||||||||||||||||||
| No. of options |
Weighted average of exercise price |
No. of options |
Weighted average of exercise price |
No. of options |
Weighted average of exercise price |
|||||||||||||||||||
| Outstanding at the beginning of the year | 45,532,659 | 0.59 | 32,888,472 | 0.65 | 40,644,792 | 0.63 | ||||||||||||||||||
| Granted | — | — | 9,586,000 | * | 0.56 | 8,050,000 | 0.57 | |||||||||||||||||
| Expired | (4,076,167 | ) | 0.6 | (1,065,305 | ) | 0.58-1.39 | (375,000 | ) | 0.6 | |||||||||||||||
| Forfeited | (4,947,135 | ) | 0.35 | (764,375 | ) | 0.6 | (1,450,000 | ) | 0.44-0.6 | |||||||||||||||
| Exercised | (3,620,885 | ) | 0.26 | — | — | - | ||||||||||||||||||
| Outstanding at the end of the year | 32,888,472 | 0.65 | 40,644,792 | 0.63 | 46,869,792 | 0.63 | ||||||||||||||||||
| Exercisable at the end of the year | 14,350,118 | 0.56 | 20,922,506 | 0.57 | 27,936,172 | 0.57 | ||||||||||||||||||
| * | Not including options to Directors and CEO which were subject to shareholder approval. |
F-47
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 14—EQUITY (continued)
The following is information about the exercise price and remaining contractual life of outstanding options:
| December 31, 2016 | December 31, 2017 | December 31, 2018 | ||||||||||||||||||||||||||||||||
| Number of outstanding options | Exercise price range in NIS | Weighted average of the remaining contractual life | Number of outstanding options | Exercise price range in NIS | Weighted average of the remaining contractual life | Number of outstanding options at the end of the year | Exercise price range in NIS | Weighted average of the remaining contractual life | ||||||||||||||||||||||||||
| 32,888,472 | 0.26 - 1.39 | 7.72 | 40,644,792 | 0.26 - 1.39 | 6.78 | 46,869,792 | 0-1.39 | 5.81 | ||||||||||||||||||||||||||
The expenses recognized in the Company’s statements of comprehensive loss in 2016, 2017 and 2018 for options granted to employees and consultants amounted to NIS 3,572 thousand, NIS 1,910 thousand and NIS 3,113 thousand, respectively.
The total unrecognized compensation cost of stock options at December 31, 2018 is approximately NIS 2.0 million. The unrecognized compensation cost of employee stock options is expected to be recognized over 4 years.
F-48
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 15—REVENUES
| A. | Revenues: |
| 2017 | 2018 | |||||||
| NIS in thousands |
||||||||
| Revenues from the sales of goods | 1,668 | 2,657 | ||||||
| Revenues from the rendering of services | - | 682 | ||||||
| Revenues from licensing agreement (see Note 2N) | - | 14,695 | ||||||
| 1,668 | 18,034 | |||||||
| B. | Revenues by geographical area (based on the location of customers): |
| 2017 | 2018 | |||||||
| NIS in thousands |
||||||||
| United states and Canada | 802 | 17,511 | ||||||
| Europe | 866 | 523 | ||||||
| 1,668 | 18,034 | |||||||
| C. | Major customers |
Set forth below is a breakdown of the Company’s revenue by major customers (major customer –revenues from these customers constitute at least 10% of total revenues in a certain year):
| 2017 | 2018 | |||||||
| NIS in thousands |
||||||||
| Customer A | 688 | 15,375 | ||||||
| Customer B | 521 | - | ||||||
| Customer C | 295 | - | ||||||
| Customer D | - | 1,816 | ||||||
F-49
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 15—REVENUES (continued)
| D. | The changes in contract liability relating to goods that were not yet delivered are as follows: |
| 2018 | ||||
| NIS in thousands |
||||
| Balance as of January 1, 2018 | - | |||
| Contract liability recognized due to LB agreement | 7,346 | |||
| Revenue recognized during the period | (37 | ) | ||
| Balance as of December 31, 2018(1) | 7,309 | |||
| Contract liability presented in current liabilities | 3,636 | |||
| Contract liability presented in non-current liabilities(2) | 3,673 | |||
| (1) | Represents the unfulfilled performance obligation related to First BioInk. |
| (2) | As of December 31, 2018, non-current contract liabilities are estimated to be recognized in 2020. |
F-50
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 16—RESEARCH AND DEVELOPMENT EXPENSES, NET
| Year ended December 31 | ||||||||||||
| 2016 | 2017 | 2018 | ||||||||||
| NIS in thousands | ||||||||||||
| Payroll and related expenses | 8,728 | 7,687 | 7,080 | |||||||||
| Share-based payments | 2,127 | 1,197 | 1,626 | |||||||||
| Subcontractors and consultants | 11,328 | 2,554 | 1,064 | |||||||||
| Consumables and materials | 1,806 | 698 | 1,112 | |||||||||
| Depreciation and amortization | 826 | 1,002 | 971 | |||||||||
| Rent and maintenance | 2,963 | 2,700 | 2,805 | |||||||||
| Other | 1,422 | 1,083 | 1,555 | |||||||||
| 29,200 | 16,921 | 16,213 | ||||||||||
| Less: | ||||||||||||
| Participation in R&D expenses, See Note 13b | (9,257 | ) | (573 | ) | - | |||||||
| IIA participation in R&D expenses, See Note 13(a)(2) | (3,154 | ) | (2,282 | ) | 3,227 | |||||||
| (12,411 | ) | (2,855 | ) | 3,227 | ||||||||
| 16,789 | 14,066 | 19,440 | ||||||||||
NOTE 17—GENERAL, ADMINISTRATIVE AND MARKETING EXPENSES
| Year ended December 31 |
||||||||||||
| 2016 | 2017 | 2018 | ||||||||||
| NIS in thousands | ||||||||||||
| Payroll and related expenses | 2,822 | 2,926 | 4,142 | |||||||||
| Share-based payments (1) | 1,445 | 2,243 | 3,514 | |||||||||
| Directors’ salary and insurance | 787 | 411 | 604 | |||||||||
| Rent and office maintenance | 364 | 321 | 321 | |||||||||
| Professional services | 5,039 | 1,827 | 3,085 | |||||||||
| Depreciation | 38 | 47 | 81 | |||||||||
| Other | 553 | 528 | 733 | |||||||||
| 11,048 | 8,303 | 12,480 | ||||||||||
| (1) | Share-based payments expenses for the year ended December 31, 2017 and 2018, include amount of NIS 1.5 million and NIS 494,000, respectively, due to fair value estimate of services received from Alpha. |
F-51
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 18—FINANCING EXPENSES (INCOME), NET
| Year ended December 31 |
||||||||||||
| 2016 | 2017 | 2018 | ||||||||||
| NIS in thousands | ||||||||||||
| Financing expenses: | ||||||||||||
| Financing expenses arising from liability to the IIA | 129 | 273 | 138 | |||||||||
| Bank and other fees | 61 | 32 | 57 | |||||||||
| Other financing expenses | - | 75 | 30 | |||||||||
| Exchange rate differences | 251 | - | - | |||||||||
| Total financing expenses | 441 | 380 | 225 | |||||||||
| Financing income: | ||||||||||||
| Remeasurement of financial instruments | - | 35 | 891 | |||||||||
| Exchange rate differences | (93 | ) | 218 | 759 | ||||||||
| Total financing income | (93 | ) | (253 | ) | (1,650 | ) | ||||||
| Financing expenses (income), net | 348 | 127 | (1,425 | ) | ||||||||
NOTE 19—LOSS PER SHARE
Basic loss per share is calculated by dividing the loss attributable to the Company’s shareholders by the weighted average number of ordinary shares issued. The calculation of the diluted loss per share did not take into account 47,944,792 options for employees and consultants, 13,745,025 Series I warrants, 36,531,500 Series K warrants, and 70,807,407 warrants, since their effect is anti-dilutive.
NOTE 20—TRANSACTIONS AND BALANCES WITH RELATED PARTIES
The Company’s key management personnel include members of the executive management and board of directors, in accordance with the definition of Related Parties in IAS 24.
| A. | Transactions with and benefits to related parties |
|
Year
ended December 31 |
||||||||||||
| 2016 | 2017 | 2018 | ||||||||||
| NIS in thousands | ||||||||||||
| CEO’s salary* | 2,010 | 1,988 | 2,129 | |||||||||
| Share-based payments portion | 1,066 | 455 | 793 | |||||||||
| Remuneration of directors | 1,214 | 613 | 1,057 | |||||||||
| Share-based payments portion | 558 | 279 | 627 | |||||||||
| Number of directors | 6 | 6 | 7 | |||||||||
Regarding benefits to other key management personnel—see C below.
| * | In accordance with the CEO’s employment agreement, the CEO will be eligible for a bonus based on qualitative criteria and parameters determined by the Company, which will amount to a maximum of four salaries, plus a special bonus based on the fulfillment of additional conditions. |
F-52
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 20—TRANSACTIONS AND BALANCES WITH RELATED PARTIES (continued)
| B. | Balances with related parties: |
| December 31 | ||||||||
| 2017 | 2018 | |||||||
| NIS in thousands | ||||||||
| For salary, incidentals and other benefits, the balance is stated in other payables under current liabilities | (577 | ) | (180 | ) | ||||
| C. | Benefits for key officers |
Compensation for the Deputy CEO & CFO, VP Research and Development, COO, VP commercialization, Chief Scientist, and VP Quality Assurance, defined as key management personnel, for their services provided to the Company, is as follows:
| Year ended December 31 |
||||||||||||
| 2016 | 2017 | 2018 | ||||||||||
| NIS in thousands | ||||||||||||
| Salary and other short-term benefits | 3,917 | 4,119 | 4,503 | |||||||||
| Share-based payments | 2,147 | 1,051 | 1,261 | |||||||||
| 6,064 | 5,170 | 5,764 | ||||||||||
| Number of key managers | 6 | 6 | 7 | * | ||||||||
| * | Includes cost of salary to two former executive officers who ended their tenure during 2018. |
F-53
CollPlant Holdings Ltd.
Notes to the Consolidated Financial Statements
NOTE 21—SUBSEQUENT EVENTS
| A. | In January 2019 Series I warrants expired without exercise. |
| B. |
On January 30, 2019, the Board of Directors appointed Jonathan M.N. Rigby as the new Chairman of the Board, effective immediately, and David Tsur stepped down from his position as Chairman and as a member of the Board.
The Company and Mr. Rigby entered into a Chairman Services Agreement (the “Agreement”), the terms of which are subject to shareholder approval. Under the Agreement, Mr. Rigby shall be entitled to an annual fee of $70,000 plus VAT, to be paid on a monthly basis and options to purchase 12,319,500 ordinary shares (represented by 246,390 ADSs exercisable at $5.07 per ADS) exercisable at $0.101 per share. The options shall have a term of seven years and shall vest upon the earlier of (1) an equity raise of at least US$10 million, in one or more financings, or (2) will vest over a period of four years, with a quarter of the options vesting on January 31, 2020, and the remaining options vesting in equal parts at the end of every quarter thereafter. |
|
In addition, the Board of Directors approved, subject to shareholder approval the grant of (i) 9,050,000 options exercisable into total of 9,050,000 ordinary shares to the company’s officers, employees and consultants, and (ii) subject to shareholders approval, 2,000,000 options exercisable into a total of 2,000,000 ordinary shares to members of the board of directors, in the following manner: (i) 250,000 options exercisable into 250,000 ordinary shares, to each of Dr. Abraham Havron, Dr. Gili Hart, Dr. Elan Penn, Scott R. Burell and Adi Goldin; and (ii) 750,000 options exercisable into 750,000 ordinary shares to Dr. Wolfgang Ruttenstorfer.
Each of the options may be exercised at a price per option of $0.101 per share. The options shall vest over a period of four years from their date of grant, with 25% of the options vesting on the first anniversary of the date of grant and the remaining options vesting equally on a quarterly basis during the three years thereafter. |
F-54